Published on January 22, 2021
EXHIBIT 10.1
EXECUTION COPY
INVESTIGATOR-INITIATED
CLINICAL RESEARCH SUPPORT AGREEMENT
This Investigator-Initiated Clinical Research Support Agreement (this “Agreement”) is made as of January 13, 2021 (“Effective Date”) by and between City of Hope National Medical Center and City of Hope Medical Foundation (collectively, “Institution”), and Lixte Biotechnology Holdings, Inc., a Delaware corporation (“Corporation”). The Institution and Corporation are each referred to herein as a “Party”, and collectively, as the “Parties”.
INTRODUCTION
This Agreement is entered into to support the research and promote an increase in the useful clinical and scientific knowledge related to the Investigator-sponsored study (the “Study”) conducted under an Institutional Review Board-approved, investigator-initiated Protocol entitled: “A Phase Ib Open-Label Study Of LB-100 In Combination With Carboplatin/Etoposide/Atezolizumab In Untreated Extensive-Stage Small Cell Lung Carcinoma, City Of Hope Protocol Number 20068” as described in Attachment A hereto (the “Protocol”, as further defined below).
AGREEMENT
In consideration of the above, and of the mutual covenants and promises contained herein and other good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, the Parties further agree as follows:
1. DEFINITIONS:
(a) “Adverse Event” means any untoward or unfavorable medical occurrence in a participant, including any abnormal sign (for example, abnormal physical exam or laboratory finding), symptom, or disease, temporally associated with the participant’s participation in the research, whether or not considered related to the participant’s participation in the research.
(b) “Corporation Information” means data, research results, formulas, technical data, and any other information relating to the Study Drug or the use thereof, which is disclosed to Institution by Corporation or its designees.
(c) “Institution Invention” any Invention that is not a Study Drug Invention.
(d) “Inventions” means all inventions (whether patentable or not), discoveries and innovations conceived, reduced to practice or made in connection with the Study or which otherwise result from the research conducted pursuant to this Agreement.
(e) “Investigator” means Ravi Salgia, M.D.
(f) “Material” means any and all Study Drug provided by Corporation to Institution hereunder together with any progeny, mutants, derivatives or parts thereof, and any materials that could not be made but for the use of the Material and/or Corporation Information. .
(g) “Protocol” means the mutually agreed protocol for the conduct of the Study as specified in Attachment A (which is incorporated herein and subject hereto), as such Protocol may amended in writing by mutual agreement of the Parties.
(h) “Serious Adverse Events” means an adverse event that results in death, is life-threatening, requires inpatient hospitalization or extends a current hospital stay, results in an ongoing or significant incapacity or interferes substantially with normal life functions, or causes a congenital anomaly or birth defect. Medical events that do not result in death, are not life-threatening, or do not require hospitalization may be considered serious adverse events if they put the participant in danger or require medical or surgical intervention to prevent one of the results listed above. “Study Drug” means the compound designated LB-100, and all analogs, metabolites and/or active forms thereof, including any derivatives or parts thereof.
(i) “Study” means the Investigator-Initiated Study conducted by Institution, in collaboration with Lixte, based on the Protocol, including all research and development work relating thereto. For the avoidance of doubt, work in connection with and furtherance of the Study and this Agreement done by Institution (whether alone or jointly with Lixte) prior to the Effective Date, including without limitation development of the Protocol, is for purposes of this Agreement deemed to be within the meaning of the term “Study” and subject to the terms of this Agreement. Without limiting the foregoing, any Confidential Information disclosed or developed in connection with such pre-Effective Date activities shall be subject to Section 9, and any Inventions developed in connection with such pre-Effective Date activities shall be subject to Section 8.
(j) “Study Data” means all data resulting from the Study or other use of the Material by Institution, including test results, clinical and/or non-clinical data, and formulation or dosage information.
(k) “Study Drug Invention” means any Invention that is comprised or consists of, or is derived from or involves the use of (including the identification or use of biomarkers related to the safety, efficacy or use of, and dosing for the treatment of small cell lung carcinoma) the Study Drug.
2. SCOPE OF WORK:
(a) Institution will be the sponsor of the Study, and Institution shall ensure that Ravi Salgia, MD is the Investigator for the Study. Institution shall comply with all obligations of a sponsor under applicable laws and regulations, and shall ensure that Investigator complies with all obligations of an investigator under applicable laws and regulations. Corporation will provide Material and funding for the Study as provided in this Agreement. No other person or entity is providing funding, or contributing proprietary drugs or materials, for this Study.
(b) Institution agrees to perform, and to cause Investigator to perform, the above titled Study in accordance with the Protocol attached to this Agreement and incorporated herein by reference. Institution shall ensure that such Study is performed in compliance with all applicable federal, state, and local statutes and regulations, with all Institutional requirements, and with all Protocol requirements, including those relating to the documentation and submission of information and reports to regulatory entities, including the FDA and Institution’s Institutional Review Board (“IRB”), all applicable privacy and data protection laws and regulations; and publications of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use including current Good Clinical Practice guidelines (“GCP”), and with this Agreement. Institution agrees and acknowledges that Corporation’s support for the Study is not being used to reward Institution’s support for any Corporation activities or to influence prescribing or formulary decisions at Institution.
(c) Institution shall ensure that all employees, consultants and agents of the Institution, the Investigator, and any participating centers where the Study is to take place (“Participating Sites”) who are assigned to perform services under this Agreement (“Project Participants”) abide by the terms of this Agreement. In performing the Study, Institution will reasonably allocate personnel with the necessary licenses, qualifications and experience to conduct the Study in accordance with the Protocol. In particular, Institution shall ensure that all Project Participants are trained in GCP. Corporation will have the right, before executing this Agreement, to review the qualifications of any key personnel, including Project Participants whose participation in the Study is expected for the duration of the Study, and raise any concerns which Corporation may have in that regard. In the event that Corporation has concerns regarding the performance of any Project Participant, the parties shall in good faith seek to resolve such concerns. Institution will notify Corporation promptly of any proposed change in Investigator. Institution will provide project-specific training to any replacement Investigator or Project Participants at its own expense. Corporation will have the right to review the qualifications of any Institution, Investigator or Project Participant replacements and raise any reasonable concerns which Corporation may have in that regard, and both parties shall seek in good faith to address such concerns.
(d) Corporation agrees to provide Institution with the quantities of Study Drug as specified in Attachment A. Institution will: (i) verify receipt of the Study Drug by signing the appropriate documentation provided by Corporation or its designee; (ii) handle and store all Study Drug securely, and as specified in the Protocol, the Study Drug labeling and/or in writing by Corporation; (iii) if instructed by Corporation in writing, over-label the Study Drug using the labeling and any specific instructions provided by Corporation, and confirm such over-labeling by promptly returning to Corporation any labeling records or documentation provided by Corporation; (iv) maintain complete and accurate records of use and disposition of the Study Drug; (v) only dispense the Study Drug to Study subjects in accordance with the Protocol; and (vi) upon completion or early termination of the Study, destroy or return to Corporation or its designee all unused Study Drug, as well as any containers (whether containing unused Study Drug or not) in accordance with Corporation’s instructions or as set forth in the Protocol, and provide Corporation with a Certificate of Destruction/Incineration to Corporation’s reasonable satisfaction.
3. TERM: The term of this Agreement will commence as of the Effective Date and will end upon the later of delivery of a final study report for the Study from Institution to Corporation or one (1) year, unless terminated earlier as provided herein.
4. PAYMENT AND SUPPORT:
(a) Corporation has agreed to provide the funding to the Institution for the Study as set forth in Attachment B. Institution agrees that the amounts payable or otherwise provided by Corporation under this Agreement represent amounts actually and reasonably required to enable the work to be performed by Institution in connection with the Study and have not been determined in a manner that takes into account the volume or value of any referrals or business. No funding or other consideration made under this Agreement is intended to be, nor shall it be construed as, an offer or payment made, whether directly or indirectly, to induce the referral of patients, the purchase, lease or order of any Corporation product, or the recommending or arranging for the purchase, lease or order of any Corporation product. Institution will maintain complete and accurate records of the use and disposition of the funding, and will make such records available to Corporation upon written request. All payments by Corporation shall be made directly to Institution, and not to any Participating Site or Investigator. Institution shall be responsible for any payments required to be made to Participating Sites or Investigator. Corporation will not be obligated to provide any quantity of Study Drug or funding other than as specified in Attachment A and B unless additional Study Drug or funding is included in a written amendment to this Agreement signed by Investigator, Institution and Corporation.
(b) Checks shall be made payable to: City of Hope National Medical Center and sent to: 1500 East Duarte Road, Duarte, California 91010, Attention: Office of Clinical Trial Support Services.
(c) Corporation shall also provide, at no charge to Institution, sufficient quantities of the Study Drug to perform the Study as described in the Protocol.
(d) Subject to Section Error! Reference source not found. (Confidentiality), Institution agrees to accurately describe Corporation’s support for the Study in accordance with any law, regulation and institutional or publication policies applicable to the activities authorized by this Agreement. Institution agrees that: (i) all claims that Institution submit for reimbursement to any national healthcare program or third party payor for any procedure that is subject to the funding provided hereunder or that involves the Study Drug provided by or on behalf of Corporation will accurately reflect the provision of such Funding or supply by or on behalf of Corporation; and (ii) Institution will not seek reimbursement from any national healthcare program or third party payor for any amounts paid, or Study Drug supplied, by Corporation under this Agreement.
5. STUDY DATA:
(a) Initiation of the Study shall not begin until the relevant approvals are obtained at Institution, and Corporation has been informed in writing of such approvals. Before submission for approval, Institution will supply Corporation with a copy of the Informed Consent Form(s) that is to be signed by all subjects enrolled in the Study (as well as all amendments thereto) (the “Consent Document”) for Corporation’s review and approval. Institution shall inform Corporation in writing of the Institutional committees’ continuing reviews of the Study promptly after each such review takes place.
(b) Institution will ensure that Corporation is named in the Consent Document, as parties to whom Study subjects’ personal data (as that term is defined in the General Data Protection Regulation (GDPR)) (“Personal Data”) and Study Data may be disclosed in connection with the Study, and that such Consent Document will permit Corporation and its designees access to Study subjects’ Personal Data via Institution as may be necessary to audit the Study via the Institution and to use the Study Data and Biological Samples (as defined below), including for research, regulatory submissions and drug development purposes. “Biological Samples” means blood, fluid and/or tissue samples collected from Study subjects as may be set forth in the Protocol, and tangible materials directly or indirectly derived from such samples. Institution will collect, retain and/or use Biological Samples solely as set forth in the Protocol.
(c) Institution shall ensure that all Study Data is recorded in a timely, accurate, complete and legible manner. Institution will take all reasonable and customary precautions, including periodic backup of computer files, to prevent the loss or alteration of such Study Data.
6. PROTOCOL AND CONSENT DOCUMENT CHANGES: Institution will not make any changes to the Protocol or the Consent Document without first informing Corporation of any such change and obtaining the written approval of the Institutional Committee and of Corporation and, if necessary, making additional regulatory submission and/or modifying regulatory submissions. Institution agrees to immediately incorporate into the Consent Document and Protocol any new core safety data provided by Corporation and to promptly seek or procure approval of the Institutional Committee for such revised Consent Document. Institution will be responsible for providing Corporation with a copy of the final Protocol and Consent Document approved by the Institutional Committee. The Protocol will be considered final after it is approved by the Institutional Committee.
7. OBLIGATIONS OF INSTITUTION:
(a) Institution will ensure that all personnel conducting the Study (i) are qualified to conduct the Study; (ii) are subject to confidentiality obligations substantially similar to those contained in this Agreement; (iii) are subject to a written agreement obligating them to assign ownership to Institution of any rights they might have in the results of their work; and (iv) will do so under the direction of Institution with the prior approval and ongoing review of all appropriate and necessary review authorities.
(b) Institution will ensure that Institution, and Study Personnel (defined below): (i) are under no contractual or other obligation or restriction that is inconsistent with Institution’s performance of or obligations under this Agreement; (ii) do not have a financial or other interest in Corporation or the outcome of the Study that might interfere with their independent judgment; (iii) are not currently: (1) debarred from providing services; (2) excluded, debarred or suspended from, or otherwise ineligible to participate in any national or state health care programs; (3) disqualified by any government or regulatory agencies from performing specific services, and are not subject to a pending disqualification proceeding; or (4) convicted of a criminal offense related to the provision of health care items or services, or under investigation or subject to any such action that is pending. During the Study and for a period of two (2) years following completion or early termination of the Study, Institution will promptly notify Corporation if Institution or, or Study Personnel are subject to the foregoing. “Study Personnel” means, collectively, Investigator, members of the Study team designated and supervised by the Investigator, on behalf of Institution, to perform Study-related procedures and/or to make Study-related decisions (“Sub-investigators”), and other Study Personnel.
(c) In accordance with GCP and applicable local regulations, Institution is responsible for assessing all clinical safety information obtained during the Study in order to produce all necessary safety reports. Institution will report all Adverse Events to the applicable regulatory authorities and the appropriate Institutional Review Board (“IRB”) as required by the Protocol, applicable law or regulation within the requisite applicable timeframes. Institution will conduct follow-up on Adverse Events as required by the Protocol, applicable law or regulation. Institution will report Serious Adverse Events requiring expedited reporting to applicable regulatory authorities via a FDA Form 3500 MEDWATCH report and concurrently provide a copy of such MEDWATCH report to Corporation. For expedited reports, Institution will send the MEDWATCH report to Corporation no later than seven (7) days for initial life-threatening and death reports, and fifteen (15) days for all other initial or follow-up serious and unexpected suspected adverse reaction (SUSAR), from the time of receipt of the SAE by Investigator and Institution. For non-expedited reports (i.e., unrelated to study drugs or a listed/expected event), Institution will send the MEDWATCH report to Corporation no later than thirty (30) days from the time of receipt of the SAE by Institution. Institution shall send all MEDWATCH reports to Corporation via email to Corporation’s regulatory group.
(d) Institution will (i) notify Corporation of any communications from or to any regulatory authority having a direct impact on the Study; (ii) include Corporation in any discussions or meetings with a regulatory authority regarding the Study where appropriate; (iii) supply Corporation with a copy of any correspondence from a regulatory authority regarding the Study, including any approval letter, and any other Study related correspondence; and (iv) allow Corporation a reasonable opportunity to comment on any correspondence being sent to the regulatory authority by Institution regarding the Study, including any submitted annual reports.
8. INTELLECTUAL PROPERTY:
(a) Corporation retains title and all rights in and to all Material and Corporation Information provided by Corporation to Institution. Institution shall have no rights in any such Material or Corporation Information, except for the limited use rights expressly granted herein. Corporation hereby grants to Institution a limited, non-exclusive, revocable, non-sublicensable and non-transferable right to use the Material and Corporation Information, solely during the term hereof, and solely internally at Institution for scientific or academic research in connection with Study conducted pursuant to the Protocol. Other than the rights to use the Materials and Corporation Information expressly granted herein, no rights are granted to Institution to any other intellectual property rights (including any patents, trade secret, trademarks or copyrights) owned by Corporation or any other party; no rights are granted hereunder by implication or estoppel; and all rights not expressly granted herein are reserved to Corporation.
(b) Institution will disclose promptly to Corporation in writing any and all Study Data and Inventions.
(c) All rights, title and interest in Institution Inventions shall be the sole and exclusive property of Institution if invented solely by Institution, and jointly owned by Institution and Corporation if jointly developed by Institution and Corporation, subject only to those rights of Corporation expressly provided herein. Corporation owns all rights, title and interest in any Study Drug Inventions; subject to the publication rights of Institution as described in Section 11, and the use rights granted to Institution in this Section 5(c). Institution hereby assigns to Corporation all rights, title and interests in and to the Study Drug Inventions. Corporation hereby grants to Institution the non-exclusive, non-transferable right to use the Study Drug Inventions solely for internal non-commercial academic or scientific research and patient care purposes.
(d) Other than the funding payments specified herein, Corporation is not obligated to make any payments to Institution in consideration of the rights and licenses granted to Corporation hereunder.
(e) Institution grants to Corporation a worldwide, royalty-free, paid-up, perpetual non-exclusive license to make or use any Institution Invention for research or development under all rights, title and interest which Institution may have or obtain in any Institution Invention.
(f) Following written notice to Corporation of an Institution Invention, Corporation shall have sixty (60) days to exercise an exclusive right of first negotiation (an “ROFN”) to obtain an exclusive, worldwide, royalty-bearing license to all rights, title and interest which Institution may have or obtain in such Invention, subject to (i) the royalty-free right of Institution and its affiliates to practice such Invention for educational and research purposes, (ii) the right of Institution and its affiliates to publicly disclose research results (subject to Section 11), and (iii) the right of Institution and its affiliates to allow other collaborators to use such Inventions for the same purposes as (i) and (ii). If Corporation exercises its ROFN by providing a written notice to Institution within sixty (60) days after receipt of written disclosure of any Invention, Corporation shall have one hundred eighty (180) days thereafter (the “Negotiation Period”) to reach agreement with Institution on license terms. If Corporation does not exercise its ROFN or if Corporation exercises its ROFN but the Parties do not complete the execution of the subject license within the Negotiation Period, then Institution may license or practice its interest in any such Inventions without the consent of and without accounting to Corporation, and Institution shall have no further obligation to Corporation whatsoever with respect thereto, subject to the non-exclusive license granted to Corporation under this Section 8(f).
(g) Institution acknowledges that except to the extent needed to exercise any rights granted to, or to perform any obligations required of Institution under this Agreement, this Agreement does not grant to the Institution any rights under any Corporation patents or any rights to use the Study Drug for commercial purpose.
9. CONFIDENTIAL INFORMATION:
(a) For purposes of this Agreement, the term “Confidential Information” shall mean all written information relating to the Study, including but not limited to data; know-how; technical and nontechnical materials; and compound samples and specifications, which a Party may disclose (the “Disclosing Party”) to the other Party (the “Receiving Party”) pursuant to this Agreement. A Disclosing Party shall use reasonable efforts to: clearly mark “confidential” any information disclosed in tangible form and considered by a Disclosing Party to be confidential or, if orally disclosed, to describe as confidential when disclosed and reduce to writing within a reasonable period of time after disclosure and marked “confidential.” Notwithstanding anything to the contrary herein, the Study Drug and Corporation Information are the Confidential Information of Corporation.
(b) Confidentiality: Each Receiving Party agrees to maintain the Disclosing Party’s Confidential Information in confidence with the same degree of care it holds its own confidential information. Neither Receiving Party will use the Disclosing Party’s Confidential Information except for the Study. Each Receiving Party will disclose the Confidential Information only to its officers, consultants and employees directly concerned with the Study, and will not disclose information to any third party not involved in the Study nor use the Confidential Information for any other purpose.
(c) Exceptions to Confidentiality: Each Receiving Party’s obligation of nondisclosure and the limitations upon the right to use the Confidential Information shall not apply to the extent that the Receiving Party can demonstrate that the information: (a) is now, or hereafter becomes, through no act or failure to act on the part of the Receiving Party, generally known or available to the public; (b) was known by the Receiving Party before receiving the information from the Disclosing Party; (c) is hereafter rightfully obtained by the Receiving Party from a third party, without breach by the third party of any obligation to the Disclosing Party; or (d) is independently developed by the Receiving Party without use of or reference to the Confidential Information by persons who had no access to the Confidential Information. Each Receiving Party may disclose the Disclosing Party’s Confidential Information if and to the extent that a disclosure is required by applicable law, provided that the Receiving Party uses reasonable efforts to limit the disclosure by means of a protective order or a request for confidential treatment and provides the Disclosing Party a reasonable opportunity to review the disclosure before it is made and to interpose its own objection to the disclosure.
(d) Survival: All obligations regarding Confidential Information under this Agreement shall survive the termination of this Agreement for a period of five (5) years; provided, however, that the obligations set forth in Section 6(e) below shall survive termination of this Agreement indefinitely.
(e) Corporation will take appropriate measures to protect the confidentiality and security of all protected health information (as such term is defined in the Health Insurance Portability and Accountability Act) that it receives from Institution in connection with the Study. If, in connection with the Study or performance of this Agreement, Corporation comes into contact with individually identifiable health information relating to patients who are not Study subjects, Corporation agrees to maintain the confidentiality of such information and not to use it for any purpose. If Corporation is permitted to receive any individually identifiable information of Study subjects under the applicable informed consent form, Corporation shall only use and disclose such information as necessary for the Study and shall promptly notify Institution of any unauthorized use or disclosure. The obligations in this paragraph shall survive the termination of this Agreement indefinitely.
10. USE OF STUDY DATA: Corporation acknowledges that Institution owns the Study Data; provided, that Corporation is hereby granted an irrevocable, perpetual royalty-free right to receive and use all Study Data, except for the disclosure of subject-identifying information, for any business purpose it deems appropriate, including, without limitation, for submission to any governmental or regulatory agencies, domestic or foreign. Institution shall promptly disclose to Corporation all Study Data.
11. REPORTS: Institution will maintain complete and up-to-date medical and other records relating to the Study and will keep Corporation informed of any Study Data and status through written reports, as reasonably requested by Corporation but no less than on a monthly basis. Institution shall furnish to Corporation a comprehensive written report within thirty (30) days after completion or early termination of the Study. At mutually agreeable times, Institution will give Corporation and its designees access to all records and documentation (however stored) relating to the Study or to the care of Study subjects, in order for Corporation to monitor the Study for source document verification and/or audit purposes. Institution will also make those records and documents available for the purposes of any audit by a regulatory authority and agree not to destroy those records and documents without first giving Corporation written notice and the opportunity to store them at Corporation’s expense. Investigator and Institution are free to publish the results of the Study, subject to the provisions in Section 11 (Publication), and to use data generated from the Study for their own research, clinical and educational purposes and programs.
12. PUBLICATION: Institution and Corporation recognize the traditional freedom of all scientists to publish and present promptly the results of their research. Institution and Corporation also recognize that patent rights can be jeopardized by public disclosure prior to the filing of suitable patent applications and that confidential information can be inadvertently disclosed. Therefore, Institution will assure that all proposed publications (including abstracts) arising from research under this Agreement will be submitted to Corporation promptly and before submission to a publisher for review. Corporation shall have thirty (30) days in which to review the publication, which shall be extended for an additional sixty (60) days when Corporation discloses reasonable need for such extension in order to file for patent protection. At Corporation’s request, Institution will delete any Corporation Confidential Information from a proposed publication.
13. INDEMNIFICATION:
(a) Institution shall indemnify and hold Corporation and its directors, officers, agents, contractors and employees harmless from any claim, liability, loss or demand by a third party arising from: (i) the negligence, recklessness or willful misconduct of Institution or any of its agents, contractors or employees, (ii) Institution’s or any of its agents’, contractors’ or employees’ failure to comply with the Protocol or any applicable law or regulations.
(b) Corporation agrees to indemnify and hold Institution, its affiliates, and their respective directors, officers, agents, contractors and employees, harmless from any claim, liability, loss or demand by a third party and arising from (i) Corporation’s use of the results of the Study Data, (ii) any defect in the Study Drug or other material supplied by Corporation for use in the Study, (iii) the negligence, recklessness or willful misconduct of Corporation or any of its agents, contractors or employees, and (iv) Corporation’s or any of its agents’, contractors’ or employees’ failure to comply with any applicable law or regulations. Notwithstanding anything to the contrary herein, Corporation shall not have any indemnity obligation under this Section 13(b) to the extent the claim, liability, loss or demand which arises from Institution’s breach of this Agreement, gross negligence or willful misconduct.
(c) The obligations of each Party under this Section are subject to: prompt notification to the indemnifying party by the indemnified party of any claim or suit (but a delay in notice excuse an indemnitor’s obligations hereunder only if and to the extent the indemnitor can show its was prejudiced by such delay); full control by the indemnifying party of any disposition or settlement of said claim or suit; and cooperation by the indemnified party with the indemnifying party regarding such disposition or settlement; provided, however, that, without the indemnified party’s prior written approval (such approval not to be unreasonably withheld), the indemnifying party shall not settle or compromise any such claim or suit if such settlement or compromise would result in an admission of liability or wrongdoing or impose any obligation on the indemnified party.
14. ENTIRE AGREEMENT: This Agreement, including any exhibits and appendices attached hereto, sets forth the entire agreement between Corporation and Institution as to its subject matter, and supersedes any and all other discussions, negotiations and representations of any kind by and among the Parties. None of the terms of this Agreement shall be amended except in writing signed by both Parties; provided, however, that the Protocol may be amended by Institution as reasonably necessary. Institution shall promptly provide to Corporation a copy of any Protocol amendment. If there is any conflict between the provisions of the final study Protocol, as it may be amended, and those of this Agreement, the provisions of this Agreement shall govern; provided, however, that the provisions of the Protocol shall govern with respect to the performance of the Study.
15. TERMINATION:
(a) If any party breaches this Agreement, the other Party may terminate it if the breaching Party does not cure the breach within thirty (30) days of written notice to the breaching Party of the same. The right of termination shall be in addition to any other rights the terminating party may have, at law or equity, pursuant to this Agreement.
(b) Each Party reserves the right to terminate this Agreement at any time effective immediately (i) if the authorization and approval to conduct the Study is withdrawn by the FDA, IRB, or other regulatory authority, or (ii) for safety or efficacy concerns.
(c) Upon receipt of notice of termination, (i) the Institution agrees to promptly terminate conduct of the Study and return any unused Study Drug and other material, if any, provided by Corporation, if applicable, and (ii) if terminated by Corporation other than for cause, will reimburse the Institution for all reasonable costs and non-cancelable commitments properly and actually incurred prior to termination in the performance of the Study consistent with this Agreement.
16. NOTICES: All notices or other communications that are required or permitted hereunder shall be in writing and delivered personally, sent by a nationally-recognized overnight courier or sent by registered or certified mail, postage prepaid, return receipt requested, to the addresses listed below or to such other addresses as each of the Parties may otherwise request. Any such communication shall be deemed to have been given (i) when delivered, if personally delivered on a business day, (ii) on the business day after dispatch, if sent by nationally-recognized overnight courier, and (iii) on the fifth business day following the date of mailing, if sent by mail.
| If to Corporation: | Lixte Biotechnology Holdings, Inc. |
| 248 Route 25A, No. 2 | |
| East Setauket, NY 11733, USA | |
| Attn: John S. Kovach, M.D. | |
| Tel: 646.894.3135 |
| If to Institution for contract or administrative matters: | |
| City of Hope National Medical Center | |
| 1500 East Duarte Road | |
| Duarte, California 91010 | |
| Attn: Office of Clinical Trial Support Services | |
| Email: CTSS-E@coh.org | |
| If to Investigator for clinical or technical matters: | |
| Ravi Salgia, MD | |
| 1500 East Duarte Road | |
| Duarte, California 91010 | |
| Email: rsalgia@coh.org |
17. RELATIONSHIP OF THE PARTIES: The execution of this Agreement shall not confer upon the Parties any interest or benefits other than those specifically set forth herein. In making and performing this Agreement, the Parties shall act at all times as independent entities and nothing contained in this Agreement shall be construed or implied to create an agency, partnership or employer and employee relationship between Corporation and Institution, Investigator, or Institution’s officers, employees, consultants or agents. Except as specifically provided herein, at no time shall either Party make commitments or incur any charges or expenses for or in the name of the other Party.
18. INDEPENDENT RESEARCH: Nothing in this Agreement shall be construed to limit the freedom of Institution or Investigator or other individuals participating in this Study, whether paid under this Agreement or not, to engage in research similar or competitive to the Study independently under other grants, contracts or agreements with parties other than Corporation.
19. SURVIVAL: Expiration or termination of this Agreement by any Party shall not affect the rights and obligations of the Parties accrued prior to the effective date of the expiration or termination. The provisions of Sections 1, 4, 5(c), 7-30 shall survive the termination or expiration of this Agreement for any reason.
20. COMPLIANCE WITH LAWS: All parties shall comply in all material respects with the requirements of all applicable laws, rules, regulations and orders of any government authority in performing the Study including, without limitation, all U.S. Food and Drug Administration regulations relating to Good Clinical Practice and clinical trials.
21. HUMAN SUBJECTS RESEARCH PROTECTION: In the event of a research injury, Institution will make medical care available to Study subjects, when appropriate, as further set forth in the informed Consent Document approved by the IRB for this Study. Corporation will report to Institution any new or unexpected Study Drug developments or information that may pose a significant health or safety risk to Study participants.
22. REPRESENTATIONS AND WARRANTIES: The Institution and Corporation each represents and warrants that (i) it is a corporation duly organized, validly existing and in good standing under the laws of its state of incorporation; (ii) it has the right and authority to execute and deliver this Agreement and to consummate the transactions contemplated hereunder; (iii) this Agreement is a legal, valid and binding agreement of the party and enforceable against it; (iv) the execution and delivery of this Agreement will not, to each party’s knowledge, violate any statute, regulation or any other restriction upon the party; and (v) it has secured all requisite authorizations and approvals necessary for the execution, delivery and performance of this Agreement. EXCEPT AS EXPRESSLY PROVIDED HEREIN, ALL MATERIAL, INFORMATION, STUDY DATA AND INVENTIONS PROVIDED, SUBMITTED OR GENERATED HEREUNDER BY THE INSTITUTION OR ITS PERSONNEL (INCLUDING WITHOUT LIMITATION THE INVESTIGATOR) IS PROVIDED, SUBMITTED OR GENERATED, AS APPLICABLE, “AS-IS” WITH NO WARRANTY OF ANY KIND, AND ALL SUCH WARRANTIES THEREIN, WHETHER STATUTORY, EXPRESS OR IMPLIED (AND INCLUDING WITHOUT LIMITATION WARRANTIES OF FITNESS FOR A PARTICULAR PURPOSE, MERCHANTABILITY, TITLE AND NON-INFRINGEMENT OF THIRD PARTY RIGHTS), ARE HEREBY DISCLAIMED TO THE MAXIMUM EXTENT PERMISSIBLE BY LAW. THE PARTIES ACKNOWLEDGE THAT THE STUDY IS EXPERIMENTAL AND THE INSTITUTION DISCLAIMS ANY WARRANTY THAT IT WILL BE ABLE TO COMPLETE THE STUDY AS CONTEMPLATED BY THE PROTOCOL OR THAT THE STUDY WILL BE SUCCESSFUL. EXCEPT WITH RESPECT TO ANY INDEMNIFICATION OBLIGATIONS OF INSTITUTION AS SET FORTH IN THIS SECTION, AND EXCEPT FOR A BREACH OF CONFIDENTIALITY, (I) THE INSTITUTION SHALL HAVE NO LIABILITY TO CORPORATION FOR ANY LOST PROFITS, LOST OPPORTUNITIES, OR CONSEQUENTIAL, SPECIAL, INCIDENTAL, INDIRECT OR PUNITIVE DAMAGES, AND (II) THE INSTITUTION’S MAXIMUM LIABILITY TO CORPORATION SHALL NOT EXCEED THE AMOUNTS PAID BY CORPORATION TO THE INSTITUTION UNDER THIS AGREEMENT.
23. DEBARMENT: Corporation hereby certifies to Institution under penalty of perjury, that Corporation has not been convicted of a criminal offense related to health care and is not currently debarred, excluded or otherwise ineligible for participation in federally funded health care programs. Corporation agrees to notify Institution in writing immediately of any threatened, proposed or actual conviction relating to health care, or any threatened, proposed or actual debarment or exclusion from participation in federally funded health care programs, of the Corporation. Corporation will not employ or contract with individuals or entities excluded from participation in a federally funded program. Any breach of this section of this Agreement by Corporation shall be grounds for immediate termination of this Agreement by Institution.
24. PUBLICITY: Neither Party shall use the other Party’s name, nor issue any public statement about this Agreement or the Study, without the prior written permission of the other Party (which permission shall not be unreasonably withheld), except as required by law (and, in such case, only with prior notice to the other Party); provided, however that Institution has the right to list the Study name and information on its Clinical Trials Online (CTOL) website system and, in order for the Institution to satisfy its reporting obligations, it may disclose the amount of support received from Corporation for the Study.
25. ASSIGNMENT: This Agreement and all rights and obligations hereunder are personal to the Parties and may not be assigned without the express written consent of the other Party, which consent will not be unreasonably withheld or delayed, provided that Corporation may assign this Agreement in its entirety in connection with the sale of all or substantially all of the business of Corporation to which this Agreement relates, including the sale of all or substantially all of Corporation’s equity or relevant, assets; a merger of Corporation (including by operation of law), whether or not Corporation is the surviving entity of such merger; or a reorganization of Corporation.
26. CHOICE OF LAW AND JURISDICTION: This Agreement shall be construed in accordance with the laws of the State of California. All actions arising under this Agreement shall be brought exclusively in the state and federal courts sitting in Los Angeles County, California and each of the Parties hereby agrees to submit to the exclusive venue and personal jurisdiction of such courts, provided that a claim for preliminary injunctive relief may be sought in any applicable jurisdiction.
27. FORCE MAJEURE: Failure of either Party to perform its obligations under this Agreement (except the obligation to make payments) shall not subject such Party to any liability or place such Party in breach of any term or condition of this agreement to the other Party if such failure is the result of any event beyond the reasonable control of such nonperforming Party, including without limitation acts of God, fire, explosion, flood, pandemic, drought, war, riot, sabotage, embargo, strike or other labor trouble, failure in whole or in part of suppliers to deliver on schedule materials, equipment or machinery, interruption of or delay in transportation, a national health emergency or compliance with any order or regulation of any government entity acting with color of right provided that: (1) the non-complying party uses reasonable efforts to cure or mitigate the effect of such force majeure event and to perform such obligation(s); (2) that party promptly (but in any event within ten (10) days of the occurrence of such event) provides written notice to the other party of the occurrence of such force majeure event, its effect on performance, and how long that party expects it to last, and thereafter provides notice(s) updating such information as reasonably necessary. For the avoidance of doubt, an increase in prices or costs, or other change in general economic conditions, or a party not having sufficient funds to comply with an obligation to pay money is not a force majeure event.
28. Waiver: The failure of a Party to enforce any breach or provision of this Agreement shall not constitute a continuing waiver of such breach or provision and such Party may at any time thereafter act upon or enforce such breach or provisions of this Agreement. Any waiver of breach executed by either Party must be in writing and shall affect only the specific breach and shall not operate as a waiver of any subsequent or preceding breach.
29. Further Instruments and Acts: Each Party shall execute and deliver such further instruments and do such further acts and things as reasonably may be required to carry out the intent and purpose of this Agreement.
30. SEVERABILITY: If any clause or provision of this Agreement is declared invalid or unenforceable by a court of competent jurisdiction or an arbitrator, such provision shall be severed and the remaining provisions of the Agreement shall continue in full force and effect. The Parties shall use their best efforts to agree upon a valid and enforceable provision as a substitute for the severed provision, taking into account the intent of this Agreement.
31. COUNTERPARTS: This Agreement may be executed in any number of counterparts, each of which shall be an original as against the Party whose signature appears thereon, but all of which taken together shall constitute but one and the same instrument.
IN WITNESS WHEREOF, the Parties have caused this Agreement to be executed by duly authorized representatives as of the Effective Date.
| Corporation | Institution | |||
| By: | By: | |||
| Name: | Name: | |||
| Title: | Title: | |||
As Investigator to this Agreement, I attest that I have read the Agreement in its entirety, and that I consent to the terms herein:
| Investigator | ||
| By: | ||
| Name: | ||
ATTACHMENT
A
PROTOCOL
[See attached City Of Hope Protocol Number 20068, Version 01]
ATTACHMENT B
Study Budget and Payment Terms
| Protocol Number: | 20068 |
| Investigator: | Ravi Salgia, MD |
| Maximum Expected Enrollment: | 42 |
| Corporation: | Lixte Biotechnology Holdings, Inc. |
| 248 Route 25A No. 2 | |
| East Setauket, NY 11733 | |
| Attention: John S. Kovach, MD | |
| Institution: | City of Hope National Medical Center |
| 1500 East Duarte Road | |
| Duarte, California 91010 | |
| Attn: Clinical Trials Support Services | |
| Tax ID Number: | 95-1683875 |
| Invoicing: | Submit all invoices under this Agreement to: |
| Lixte Biotechnology Holdings, Inc. | |
| 248 Route 25A No. 2 | |
| East Setauket, NY 11733 | |
| Attention: Eric Forman | |
| 646.894.3135 | |
| ap@lixte.com, eforman@lixte.com |
The total cost will be paid by the Corporation in U.S. dollars.
The total cost ($2,958,210.07) will be paid by the Corporation to Institution in installments according to the following schedule:
| 1. | $240,508.00 | Nonrefundable, upon execution of Agreement |
| 2. | $285,019.78 | Within 30 days of Study activation |
| 3. | $285,019.78 | Upon enrollment of 5 patients |
| 4. | $285,019.78 | Upon enrollment of 10 patients |
| 5. | $285,019.78 | Upon enrollment of 15 patients |
| 6. | $285,019.78 | Upon enrollment of 20 patients |
| 7. | $285,019.78 | Upon enrollment of 25 patients |
| 8. | $285,019.78 | Upon enrollment of 30 patients |
| 9. | $285,019.78 | Upon enrollment of 35 patients |
| 10. | $285,019.78 | Upon enrollment of 40 patients |
| 11. | $114,007.91 | Upon enrollment of 42 patients |
| 12. | $147,910.50 | Upon receipt of a Final Study Report. This payment is not dependent on the number of patients enrolled, only the receipt of a Final Study Report. |
Notwithstanding anything to the contrary in the Investigator-Initiated Clinical Research Support Agreement, only payment no. 1 is non-refundable, and any other amounts paid in advance will be refunded in the event patients are not enrolled or if the trial does not otherwise proceed, subject to the last sentence of this paragraph. Payment no. 2 will be refunded in its entirety if no patient is enrolled within ninety (90) days of Study activation. In case Institution does not reach an enrollment milestone (payments 3 through 11), Corporation agrees to prorate Institution’s expenses and agrees to pay for enrolled patients at a rate of $69,001.46 per patient. Should Institution reasonably determine and can reasonably demonstrate that the payments do not adequately compensate the Institution for services rendered, the Institution may request that Corporation consider a modification in payments. Corporation shall consider such request in good faith.
All payments will be due within thirty (30) days following receipt by the Corporation of an invoice from Institution.
| Page 1 of 92 |
(i) CITY OF HOPE NATIONAL MEDICAL CENTER
1500 E. DUARTE ROAD
DUARTE, CA 91010
DEPARTMENT OF MEDICAL ONCOLOGY AND THERAPEUTICS RESEARCH
| TITLE: | A PHASE Ib OPEN-LABEL STUDY OF LB-100 IN COMBINATION WITH |
| CARBOPLATIN/ETOPOSIDE/ATEZOLIZUMAB IN UNTREATED EXTENSIVE-STAGE SMALL CELL LUNG CARCINOMA |
| CITY OF HOPE PROTOCOL NUMBER: | 20068 | VERSION: | 01 |
| SPONSOR/IND NUMBER: | City of Hope/IND 151424 |
| DISEASE SITE: | Lung |
| STAGE (if applicable): | Extensive-stage |
| MODALITY: | Combined immunotherapy and chemotherapy |
| PHASE/TYPE: | Phase Ib |
| PRINCIPAL INVESTIGATOR: | Ravi Salgia, MD, PhD |
| COLLABORATING INVESTIGATOR(S): | Vincent Chung, M.D. |
|
Marianna Koczywas, M.D. Erminia Massarelli, M.D. John Kovach, M.D. |
|
| (ii) PARTICIPATING CLINICIANS: | |
| PARTICIPATING SITES: | City of Hope |
| INDUSTRY PARTNER: | Lixte Biotechnology Holdings, Inc. |
| AGENT NSC# AND IND#: | NSC D753810, IND 109777 |
| COORDINATING CENTER: | City of Hope Comprehensive Cancer Center |
| Page 2 of 92 |
| II. | Protocol Team |
| (i) | PRINCIPAL INVESTIGATOR: | |
| BIOSTATISTICIAN | ||
| Ravi Salgia, Professor and Chair | Paul Frankel, PhD | |
| Department of Medical Oncology and Therapeutics | Division of Biostatistics | |
| Research | ||
| 1500 E Duarte Ave. | 1500 E Duarte Ave. | |
| Duarte, CA 91010 | Duarte, CA 91010 | |
| Phone: 626-218-3712 | Phone: 626-218-5265 | |
| Fax: 626-471-7322 | e-mail: pfrankel@coh.org | |
| e-mail: rsalgia@coh.org | ||
| (ii) COLLABORATING INVESTIGATORS: | ||
| Vincent Chung, M.D. | Marianna Koczywas, M.D. | |
| Department of Medical Oncology and Therapeutics | Department of Medical Oncology and Therapeutics | |
| Research | Research | |
| 1500 E Duarte Ave | 1500 E Duarte Ave. | |
| Duarte, CA 91010 | Duarte, CA 91010 | |
| Phone: 626-218-3712 | Phone: 626-218-3712 | |
| Fax: 626-471-7322 | Fax: 626-471-7322 | |
| e-mail: vchung@coh.org | e-mail: MKoczywas@coh.org | |
| Erminia Massarelli, M.D. | John Kovach, M.D. | |
| Department of Medical Oncology and Therapeutics | Lixte Biotechnology Holdings, Inc | |
| Research | 248 Route 25A No. 2. | |
| 1500 E Duarte Ave. | East Setauket, NY 11733 | |
| Duarte, CA 91010 | Phone: 631-880-2907 | |
| Phone: 626-218-3712 | Fax: 631-982-5050 | |
| Fax: 626-471-7322 | e-mail: jkovach@lixte.com | |
| e-mail: emassarelli@coh.org | ||
| (iii) PROTOCOL DEVELOPMENT SCIENTIST: | ||
| Tim Synold, Pharm.D. | Sarah Cole | |
| Beckman Research Institute | Clinical Protocol Development Center | |
| 1500 E Duarte Ave. | 1500 E Duarte Ave. | |
| Duarte, CA 91010 | Duarte, CA | |
| Phone: 626-218-1110 | Phone: 626-218-0719 | |
| e-mail: tsynold@coh.org | e-mail: scole@coh.org |
| Page 3 of 92 |
Study Schema
| Page 4 of 92 |
Protocol Synopsis
Protocol Title:
A Phase Ib Open-Label Study of LB-100 in combination with Carboplatin/Etoposide/Atezolizumab in Untreated Extensive-Stage Small Cell Lung Carcinoma
Rationale for this Study:
More than one million people died from lung cancer worldwide in 2017, and small cell carcinomas account for approximately 15% of all lung cancers. Even with double or triple drug therapy combinations, median survival for SCLC with “extensive disease” (ED-SCLC, 70% of patients) is only approximately 9 months and overall 5-year survival remains at around 5%. PP2A is ubiquitously expressed in SCLC cells (unpublished data), however, its potential relevance in SCLC remains mostly unknown. Protein phosphatase 2A (PP2A) is a phosphatase involved in the regulation of key oncoproteins, such as c-Myc and Bcr-Abl in a wide range of cancer subtypes including lung cancers and B cell-derived leukemias. A recently published study by Xiao et al (Cell 2018) demonstrated that PP2A redirected glucose carbon utilization from glycolysis to the pentose phosphate pathway to salvage to oxidative stress in B-lymphoid cells. Moreover, the same mechanism was observed in SCLC cell lines with PAX5 expression. These findings suggest a previously unexplored rationale for specific targeting of PP2A in SCLC. LB-100 is a potent and selective antagonist of PP2A that has shown efficacy in a number of pre-clinical models. The combination of LB-100 with carboplatin, etoposide and atezolizumab, the standard of care for ED-SCLC, will be evaluated in treatment naïve patients to determine the recommended phase II dose (RP2D).
Objectives:
The primary objective of this study is to determine the recommended Phase II dose (RP2D) of LB-100 when given in combination with standard doses of carboplatin, etoposide and atezolizumab in treatment naïve patients with extensive-stage small cell lung cancer (ED-SCLC).
The secondary objectives of the study are:
| ● | Progression Free Survival (PFS) |
| ● | Objective response rate (ORR) |
| ● | Overall survival (OS) |
| ● | Duration of overall response (DOR) |
| ● | Safety/Adverse events |
Exploratory objectives of the study are:
| ● | The pharmacokinetics (PK) of LB-100 and etoposide |
| ● | The biomarkers relevant to LB-100 and the disease state as well as their correlation to clinical outcomes |
Study Design:
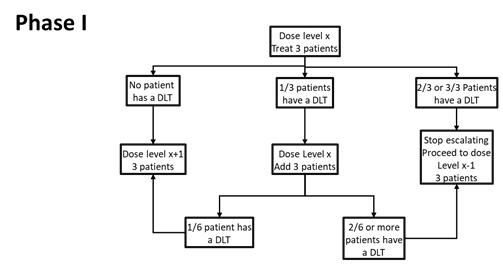
Dose Escalation: The Phase I dose-finding will use a traditional 3+3 to determine the maximum tolerated dose (MTD), based on first cycle DLTs. A maximum of 4 dose levels of LB-100 will be explored. The determination of the recommended Phase II dose (RP2D) will be based on the MTD (and will not exceed the MTD) with additional consideration of dose modifications, adverse events in subsequent cycles, clinical activity and correlative studies.
Expanded Cohort: Additional patients will be enrolled until 12 patients are treated at the proposed RP2D to help confirm the tolerability of the RP2D and obtain preliminary data on efficacy
| Page 5 of 92 |
Primary and Secondary Endpoints:
Primary endpoints:
| ● | Determine recommended phase II dose (RP2D) of the combination using DLT (Definition Section 5.7) during the first cycle as assessed by CTCAE version 5.0 |
Secondary endpoints:
| ● | Objective response rate (ORR) by RECIST v1.1 (Appendix B) | |
| ● | Duration of overall response by RECIST v1.1 (Appendix B) | |
| ● | Safety and Adverse events by assessed by CTCAE version 5.0 | |
| ● | Progression-free survival (PFS) as defined by RECIST v1.1 (Appendix B) | |
| ● | Overall survival, which is defined as the time from the date of study enrollment to the date of death from any cause. For patients who are still alive as of the data cutoff date, OS time will be censored on the date of the patient’s last contact (last contact for patients in post discontinuation is last known alive date in mortality status). |
Sample Size/Accrual/Study Duration:
Sample Size: Minimum=14, Maximum=36, Expected=21
Estimated Accrual Duration: 1-1.5 years
Estimated Study Duration: 18 -24 months
Estimated Participant Duration: 6 months
Abbreviated Eligibility Criteria:
Main Inclusion Criteria:
| ● | Histologically or cytologically confirmed extensive-stage disease small cell lung carcinoma per the Veterans Administration Lung Study Group (VALG) staging system, (Appendix E) | |
| ● | Measurable disease as defined by the Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 (Appendix B) | |
| ● | No prior systemic chemotherapy, immunotherapy, biological, hormonal, or investigational therapy for SCLC | |
| ● | Adequate hematologic and organ function, including: | |
| Hematologic: absolute neutrophil (segmented and bands) count (ANC) ≥1.5x10/L, platelets ≥100x10/L, and hemoglobin ≥9 g/dL | ||
| Hepatic: bilirubin ≤1.5 times upper limits of normal (ULN) may be enrolled, and alkaline phosphatase (AP), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) ≤3.0 times ULN (AP, AST, and ALT ≤5 times ULN are acceptable if the liver has tumor involvement). | ||
| Renal: calculated creatinine clearance (CrCl) ≥60 mL/min based on the Cockcroft and Gault formula (as defined in Section 3.3) |
| Page 6 of 92 |
Main Exclusion Criteria:
| ● | Diagnosis of NSCLC or mixed NSCLC and SCLC | |
| ● | No prior malignancy other than SCLC, carcinoma in situ of the cervix, or nonmelanoma skin cancer, unless that prior malignancy was diagnosed and definitively treated 5 or more years prior to study entry with no subsequent evidence of recurrence. Patients with a history of low grade (Gleason score ≤6=Grade Group 1) localized prostate cancer will be eligible even if diagnosed less than 5 years prior to study entry |
Investigational Product Dosage and Administration:
One Cycle is 21 Days. Patients will receive 4 cycles of induction LB-100 + atezolizumab/carboplatin/etoposide and then will proceed to maintenance with atezolizumab + LB-100.
LB-100: Intravenous (IV) at assigned dose (.83, 1.25, 1.75, 2.33 or 3.10 mg/m2), over 15 minutes, given first, Days 1 & 3 of each cycle during induction and maintenance. Other drugs should be given 1 hour after the end of the LB-100 infusion.
Atezolizumab: 1,200 mg IV after LB-100, Day 1 of each cycle during induction and maintenance. Infused over 60 (± 15) minutes (for first infusion, shortening to 30 [± 10] minutes for subsequent infusions, depending on patient tolerance), given after LB-100.
Carboplatin: 5 AUC IV, after the atezolizumab, over 30-60 minutes, Day 1 of each cycle during induction.
Etoposide: 100 mg/m2 IV, given last (after the carboplatin on Day 1 of each cycle, by itself Day 2 of each cycle, after LB-100 Day 3 of each cycle) during induction. Infused over 60 minutes.
Clinical Observations and Tests to be Performed:
Efficacy: CT/PET/MRI scans
Safety: Adverse events (AEs) by CTCAE 5.0/serious adverse events (SAEs), clinical chemistry, hematology
Bioanalytical: Blood samples to measure plasma LB-100, endothall, and etoposide concentrations Pharmacokinetic: LB-100 and etoposide exposure
Abbreviated Statistical Considerations:
Safety: All patients who receive at least one dose of study drug will be evaluated for safety and toxicity. Safety analyses will include the following: summaries of the adverse event rates (including all events and study drug-related events), all serious adverse events (SAEs), deaths on-study, deaths within 30 days of the last dose of study drug, and discontinuations from study drug due to adverse events; listings and frequency tables categorizing laboratory and nonlaboratory adverse events by maximum CTCAE 5.0 grade and relationship to study drug.
Expanded Cohort:12 patients at the RP2D will help confirm the choice of RP2D. If during the expansion cohort, more than 30% of the patients at initial RP2D experience a DLT, the study will hold accrual (accrual can also be held at the discretion of the PI for non-DLT or other safety considerations). With 12 patients, any serious treatment-related adverse event that occurs with a true frequency of 10%, will be observed at least once with a probability of 72%, and any such AE with a true frequency of 20% would be observed at least once with a probability of 93%. The DLT rate can be estimated with a standard error of at most 14%.
| Page 7 of 92 |
2.2 Table of Contents
| SECTION | PAGE |
| Protocol Team | 2 |
| Table of Contents | 7 |
| Abbreviations | 12 |
| 1.0 Goals and Objectives (Scientific Aims) | 13 |
| 2.0 Background | 13 |
| 2.1 Introduction/Rationale for Development | 13 |
| 2.1.1 Small Cell Lung Carcinoma | 14 |
| 2.1.2 Treatment of Extensive-Stage Disease Small Cell Lung Carcinoma | 15 |
| 2.1.3 PP2A | 15 |
| 2.1.4 LB-100 | 16 |
| 2.2 Rationale and Overview of Proposed Study | 16 |
| 2.3 Preclinical Toxicity Studies of LB-100 | 16 |
| 2.3.1 LB-100 in Rats | 16 |
| 2.3.2 LB-100 in Dogs | 17 |
| 2.4 Human Studies | 19 |
| 2.4.1 Single-Agent Phase I Study LB-100 | 19 |
| 3.0 Patient Eligibility | 22 |
| 3.1 Inclusion Criteria | 22 |
| 3.1.1 Disease Status | 22 |
| 3.1.2 Age Criteria, Performance Status, and Life Expectancy | 22 |
| 3.1.3 Child Bearing Potential | 22 |
| 3.1.4 Protocol-Specific Criteria | 23 |
| 3.1.5 Informed Consent/Assent | 23 |
| 3.1.6 Prior Therapy | 23 |
| 3.2 Exclusion Criteria | 23 |
| 3.2.1 Non-Compliance | 23 |
| 3.3 Inclusion of Women and Minorities | 25 |
| Page 8 of 92 |
| 4.0 Screening and Registration Procedures | 25 |
| 4.1 Pre-Enrollment Informed Consent and Screening Procedures | 25 |
| 4.2 Participant Enrollment | 25 |
| 4.2.1 COH DCC Availability and Contact Information | 25 |
| 4.2.2 Slot verification and reservation | 25 |
| 4.2.3 Registration Process | 26 |
| 4.3 Screen Failures and Registered Participants Who Do Not begin Study Treatment | 26 |
| 4.4 Screening Procedures | 26 |
| 4.5 Informed Consent | 26 |
| 4.6 Registration Requirements/Process | 26 |
| 4.7 Dose Level Assignment | 27 |
| 5.0 Treatment Program | 27 |
| 5.1 Treatment Overview | 27 |
| 5.1.1 Schedule | 27 |
| 5.2 Planned Duration of Therapy | 30 |
| 5.2.1 Baseline and Study Treatment Periods | 30 |
| 5.2.2 Postdiscontinuation Period | 31 |
| 5.3 Criteria for Removal from Treatment | 31 |
| 5.4 Subject Follow-Up | 32 |
| 5.5 Supportive Care, Other Concomitant Therapy, Prohibited Medications | 32 |
| 5.6 Additional Studies | 33 |
| 5.7 Definition of Dose-Limiting Toxicity (DLT) | 34 |
| 6.0 Dose Delays/Modifications for Adverse Events | 35 |
| 6.1 Dose Modifications | 35 |
| 6.2 Carboplatin/Etoposide Dose Modifications | 35 |
| 6.2.1 Hematologic Toxicity | 36 |
| 6.2.2 Non-Hematologic Toxicity | 36 |
| 6.3 Atezolizumab Dose Holding | 36 |
| 6.3.1 Management of Atezolizumab-Specific Adverse Events | 48 |
| 6.4 LB-100 Dose Modifications | 49 |
| 6.4.1 Hematologic Toxicity | 49 |
| 6.4.2 Non-hematologic Toxicity | 50 |
| Page 9 of 92 |
| 6.5 Pharmacokinetic Studies | 50 |
| 6.5.1 Pharmacokinetics | 50 |
| 7.0 Unanticipated Problems and Adverse Event Reporting | 52 |
| 7.1 Definitions | 52 |
| 7.1.1 Adverse Event | 52 |
| 7.1.2 Serious Adverse Event (SAE) | 53 |
| 7.1.3 Unanticipated Problems Involving Risks to Subjects or Others | 53 |
| 7.1.4 Adverse Events of Special Interest (AESI) | 54 |
| 7.2 Assessment of Adverse Events | 54 |
| 7.3 Reporting of Adverse Events | 55 |
| 7.3.2 Expediting Reporting Requirements of SAEs and UPs | 56 |
| 7.3.3.1.Reporting to the FDA | 56 |
| 7.3.3.2.Reporting to Lixte Biotechnology | 56 |
| 8.0 Agent Information and Risks | 57 |
| 8.1 LB-100 | 57 |
| 8.1.1 Description | 57 |
| 8.1.2 Pharmacology – Handling, Storage, Dispensing and Disposal | 57 |
| 8.2 Carboplatin | 57 |
| 8.2.1 Description | 57 |
| 8.2.2 Toxicology | 58 |
| 8.2.3 Pharmacology – Handling, Storage, Dispensing and Disposal | 58 |
| 8.3 VP-16 (Etoposide) | 58 |
| 8.3.1 Description | 58 |
| 8.3.2 Toxicology | 59 |
| 8.3.3 Pharmacology – Handling, Storage, Dispensing and Disposal | 59 |
| 8.4 Atezolizumab | 60 |
| 8.4.1 Description | 60 |
| 8.4.2 Toxicology | 60 |
| 8.4.3 Pharmacology – Handling, Storage, Dispensing and Disposal | 62 |
| 9.0 Correlative/Special Studies | 62 |
| 10.0 Study Calendar | 63 |
| 11.0 Endpoint Definitions | 64 |
| Page 10 of 92 |
| 12.0 Data Handling, Data Management, Record Keeping | 64 |
| 12.1 Source Documents | 64 |
| 12.2 Data Capture Methods and Management | 65 |
| 12.3 Case Report Forms/Data Submission Schedule | 65 |
| 12.4 Regulatory Records | 66 |
| 12.5 Protocol Deviations and Single Subject Exceptions | 66 |
| 12.5.1.1 Deviation | 66 |
| 13.0 Statistical Considerations | 67 |
| 13.1 Study Design | 67 |
| 13.2 Sample Size Accrual Rate | 68 |
| 13.3 Statistical Analysis Plan | 68 |
| 13.3.1 General Considerations | 68 |
| 13.3.2 Patient Disposition | 68 |
| 13.3.3 Patient Characteristics | 68 |
| 13.3.4 Concomitant Therapy | 69 |
| 13.3.5 Postdiscontinuation Therapy | 69 |
| 13.3.6 Treatment Compliance | 69 |
| 13.3.7 Primary Outcome and Methodology | 68 |
| 13.3.8 Pharmacokinetic/Pharmacodynamic Analyses | 69 |
| 13.3.9 Safety Analyses | 69 |
| 13.3.10 Interim Analyses | 69 |
| 14.0 Human Subject Issues | 70 |
| 14.1 Institutional Review Board | 70 |
| 14.2 Recruitment of Subjects | 70 |
| 14.3 Advertisements | 70 |
| 14.4 Study location and Performance Sites | 70 |
| 14.5 Confidentiality | 70 |
| 14.6 Financial Obligations and Compensation | 70 |
| 14.7 Informed Consent Processes/Regulatory Considerations | 71 |
| 14.7.1 Regulatory Considerations | 71 |
| 14.7.2 Investigator Information | 72 |
| 14.7.3 Protocol Signatures | 72 |
| 14.7.4 Final Report Signature. | 72 |
| Page 11 of 92 |
| 15.0 Study Oversight, Quality Assurance, and Data & Safety Monitoring | 72 |
| 15.1 All Investigator Responsibilities | 72 |
| 15.2 Study Principal Investigator Responsibilities | 73 |
| 15.3 Protocol Management Team (PMT) | 73 |
| 15.4 Monitoring | 73 |
| 15.5 Quality Assurance | 73 |
| 15.6 City of Hope Data and Safety Monitoring Committee | 74 |
| 16.0 References | 75 |
| Appendix A: Performance Status Scales | 79 |
| Appendix B: RECIST v1.1 Criteria | 80 |
| Appendix C: Registration Coversheet | 87 |
| Appendix D: Expedited Reporting CoverSheet | 88 |
| Appendix E: Veterans Administration Lung Study Group (VALG) Staging System for SCLC… | 89 |
| Appendix F: Common Substrates of CYP450 Isoenzymes | 90 |
| Page 12 of 92 |
| III. | Abbreviations |
| Abbreviation | Meaning | |
| AE | Adverse Event | |
| CFR | Code of Federal Regulations | |
| COH | City of Hope | |
| CR | Complete Response | |
| CRA | Clinical Research Associate | |
| CRF | Case Report Form | |
| CTCAE | Common Terminology Criteria for Adverse Events | |
| CTEP | Cancer Therapy Evaluation Program | |
| DLT | Dose Limiting Toxicity | |
| DSMC | Data Safety Monitoring Committee | |
| FDA | Food and Drug Administration | |
| GCP | Good Clinical Practice | |
| IB | Investigator Brochure | |
| ICF | Informed Consent Form | |
| IDS | Investigational Drug Services | |
| IND | Investigational New Drug | |
| IRB | Institutional Review Board | |
| MTD | Maximum Tolerated Dose | |
| NCI | National Cancer Institute | |
| PD | Progressive Disease | |
| PI | Principal Investigator | |
| PMT | Protocol Monitoring Team | |
| PR | Partial Response | |
| SAE | Serious Adverse Event | |
| SD | Stable Disease |
| Page 13 of 92 |
| IV. | 1.0 Goals and Objectives (Scientific Aims) |
1.1. Goals
This is a Phase Ib open label study for subjects with extensive-stage disease SCLC who have not received prior treatment with systemic therapy for SCLC. The Phase Ib study is a single arm study expected to enroll 18 evaluable patients (maximum 30) entered in groups of 3 at escalating doses of LB-100 using the traditional 3+3 design. Patients will receive induction therapy with carboplatin/etoposide/atezolizumab for 4 cycles. Each cycle is defined as 3 weeks (21 days). Patients will then proceed to maintenance with LB-100 and atezolizumab. Patients who discontinue study therapy without disease progression will continue to be evaluated for tumor response using RECIST v1.1 (Appendix B) guidelines every 6-8 weeks until disease progression, death, or study closure. The primary endpoint is to determine the recommended phase II dose (RP2D) of LB-100 plus carboplatin/etoposide/atezolizumab in patients with extensive-stage small cell lung carcinoma.
1.2. Objectives
1.2.1. Primary Objectives
To determine the recommended phase II dose (RP2D) of LB-100 when given in combination with standard carboplatin/etoposide/atezolizumab in treatment naïve patients with extensive-stage small cell lung cancer (ED-SCLC).
1.2.2. Secondary Objectives
The secondary objectives of the study are to assess the following variables:
| ● | Objective response rate (ORR) | |
| ● | Progression-free survival (PFS) | |
| ● | Overall survival (OS) | |
| ● | Duration of overall response (DOR) | |
| ● | Safety and adverse events (AEs) |
1.2.3. Exploratory Objectives
| ● | The pharmacokinetics (PK) of LB-100 and etoposide | |
| ● | The biomarkers relevant to LB-100 and the disease state as well as their correlation to clinical outcomes |
| 2.0 | Background | |
| 2.1 | Introduction/Rationale for Development |
2.1.1 Small Cell Lung Carcinoma
Lung cancer is the leading cause of cancer mortality worldwide, with one million new cases annually. Small cell lung cancer (SCLC) is an aggressive form of cancer that is strongly associated with cigarette smoking. In United States, in 2010, 222,000 new cases of lung cancer were diagnosed of which 35,000 were SCLC (American Cancer Society). The median age of SCLC patients is 63, and more than 25% are over the age of 70 (1). Small cell lung cancer is a rapidly growing tumor with a high rate of metastases in comparison to non-small cell lung cancer (NSCLC). Patients are staged according to a two-stage system, which was developed by the Veterans Administration Lung Cancer Study Group, consisting of limited-stage disease (LD -SCLC) or extensive-stage disease (ED-SCLC)(2). Limited-stage disease SCLC is confined to a single hemithorax region within an acceptable radiation field. Approximately 65% to 70% of patients with SCLC present with ED-SCLC, which is found beyond a hemithorax region. Untreated patients with ED-SCLC have a median survival of approximately 5 weeks; patients treated with chemotherapy have a median survival of 7 to 11 months (3). Extensive-stage disease-SCLC has a 2-year survival rate of less than 10% with current management options.
| Page 14 of 92 |
2.1.2 Treatment of Extensive-Stage Disease Small Cell Lung Carcinoma
Combination chemotherapy remains the focus of treatment for patients with ED-SCLC. In the 1970s and early 1980s, CAV (cyclophosphamide, doxorubicin, and vincristine) was the most commonly used regimen. In the mid-1980s, etoposide was discovered as an active agent in SCLC, and preclinical investigations demonstrated synergy between etoposide and cisplatin. Randomized clinical studies confirmed that this combination was as effective as CAV, with less toxicity (3).
Several other agents have been shown to have activity in SCLC, and many studies have compared 3-drug regimens to the standard 2-drug regimens with no improvement in efficacy. A Phase 3 trial conducted by the Norwegian Lung Cancer Study Group randomized 436 patients, including 214 patients with LD-SCLC and 222 patients with ED-SCLC. Patients received etoposide plus cisplatin or a combination of cyclophosphamide, epirubicin, and vincristine (CEV). Median survival for patients with ED-SCLC was 8.4 months in the etoposide plus cisplatin arm and 6.5 months in the CEV arm (p=.21) (4). In 2005, Phase 3 study conducted by the Cancer and Leukemia Group B (CALGB) compared the combination etoposide/cisplatin with or without paclitaxel and granulocyte colony-stimulating factor (G-CSF) in patients with ED-SCLC (5). A total of 565 patients were randomized. Median progression-free survival time on the carboplatin/etoposide arm was 5.9 months compared with 6 months for patients receiving carboplatin/etoposide/paclitaxel, and median overall survival was 9.9 months on the etoposide/cisplatin arm and 10.6 months on the paclitaxel arm. Toxic deaths occurred in 2.4% of the patients not receiving paclitaxel and 6.5% of patients being treated with paclitaxel. Thus, the addition of paclitaxel to etoposide and cisplatin did not improve survival and was associated with unacceptable toxicity in patients with ED-SCLC (5). Results from one of the largest studies ever conducted for patients with ED-SCLC were also reported in 2005. This study included 784 patients randomized to receive either topotecan plus cisplatin or the standard etoposide plus cisplatin; efficacy was comparably seen in overall response rates (63% versus 69%), median time to progression (24.1 versus 25.1 weeks), median survival (39.3 versus 40.3 weeks), and 1-year survival rates (31.4% for both arms) (6).
More recently the phase III IMpower133 randomized double-blind study evaluated whether adding a checkpoint inhibitor of programmed death signaling (atezolizumab) might improve chemotherapy benefits in patients with ED-SCLC (7). A total of 201 patients were randomly assigned to the platinum/etoposide/atezolizumab arm and 202 were assigned to the placebo arm. The median progression- free survival time on the platinum/etoposide arm was 4.3 months as compared with 5.2 months with platinum/etoposide/atezolizumab. The median overall survival was 12.3 months in the platinum/etoposide/atezolizumab arm and 10.3 months in the placebo group. The addition of immunotherapy to etoposide and platinum chemotherapy improved overall survival and progression-free survival and was not associated with unacceptable toxicity in patients with ED-SCLC (7). IMpower133 is considered the first study in 20 years to show a clinically meaningful improvement in overall survival over the standard of care in frontline ED-SCLC.
| Page 15 of 92 |
Carboplatin has been studied in a variety of human solid tumors (ovarian, head and neck, non-small cell lung, and small cell lung) with objective response rates between 10% and 85%. It has also been used successfully in combination with a number of other cytotoxic agents for the treatment of ovarian cancer, NSCLC, and SCLC (8-10). A 1992 review of Phase 2 and 3 studies with carboplatin in patients with SCLC determined carboplatin to be one of the most active agents in untreated SCLC (11).
Platinum-based therapy (carboplatin or cisplatin) combined with etoposide is a current standard of care for patients with ED-SCLC. However, carboplatin is often preferred over cisplatin, as it provides advantages such as fewer gastrointestinal, renal, auditory, and neurologic toxicities as well as easier administration (12).
2.1.3 PP2A
Protein phosphatase 2A (PP2A) is a ubiquitous serine/threonine phosphatase that is a master tumor suppressor involved in key regulation of oncoproteins, such as c-MYC and BCR-ABL in lung cancer and other cancer types. It has a broad range of cellular regulatory functions such as cell survival, apoptosis, mitosis, and DNA-damage response (13). Previous studies and a more recently a Phase I clinical trial have shown that PP2A inhibition can potentially sensitize tumors to radiation and chemotherapy (14). In a Phase I clinical trial of LB-100, a small molecule inhibitor of PP2A derived from natural compound cantharadin, in advanced solid tumors LB-100 was well tolerated and 10 out of 20 patients had achieved stable disease (15). Given the ubiquity of PP2A, the inhibition of LB-100 likely has multiple downstream effects. Preclinical studies indicate that PP2A inhibition with LB-100 can result in down regulation of DNA- damage response (16-18) abrogation of cell cycle checkpoint (16, 19), increase HIF dependent tumor angiogenesis (20), and induction of cellular differentiation by inhibition of N-CoR complex formation (16).
Moreover Xiao et al. 2018 showed that PP2A redirected glucose carbon utilization from glycolysis to the pentose phosphate pathway (PPP) to salvage oxidative stress, revealing a gatekeeper function of the PPP in a broad range of B cell malignancies that can be efficiently targeted by small molecule inhibition of PP2A and G6PD(21).
2.1.4 LB-100
LB-100 (3-(4methylpiperazine-carbonyl)-7-oxalobicyclo[2.2.1]heptane-2-carboxylic acid; NSC D753810) is a small molecule (MW 268), which inhibits protein phosphatase 2A (PP2A) about 80 fold more efficiently than protein phosphatase 1 (PP1). The compound has single agent activity in vitro and in vivo and potentiates the activity of cytotoxic agents including temozolomide (TMZ), doxorubicin (DOX), docetaxel, and ionizing radiation in vivo. The mechanism of potentiation appears to be inhibition of cell cycle and mitotic checkpoints induced by non-specific DNA damaging agents, allowing dormant cancer cells to enter S phase and continue in mitosis despite acute DNA damage (22). LB-100 also appears to affect the vasculature inducing transient reversible vessel “leakiness” at high doses. Because of its unique mechanism of action and ability to enhance the activity of a broad spectrum of anti-cancer agents including ionizing radiation, LB-100 has the potential to be useful for the treatment of many types of cancer as well as being the first-in-class of a new type of signal transduction modulator.
| Page 16 of 92 |
2.2 Rationale and Overview of Proposed Study
PP2A has been shown to be ubiquitously expressed in SCLC cells but not adjacent normal tissue by immunohistochemistry (unpublished data). Additionally, PP2A catalytic and structural subunits are abundantly expressed in SCLC cell lines H82, H524, H146 and H69, but weakly expressed in control HBEC 3KTcells by western blot analysis (unpublished data). Protein phosphatase 2A (PP2A) is a phosphatase involved in the regulation of key oncoproteins, such as c-Myc and Bcr-Abl in a wide range of cancer subtypes including lung cancers and B cell-derived leukemias. A recently published study by Xiao et al (Cell 2018) demonstrated that PP2A redirected glucose carbon utilization from glycolysis to the pentose phosphate pathway to salvage to oxidative stress in B-lymphoid cells. Moreover, the same mechanism was observed in SCLC cell lines with PAX5 expression. These findings suggest a previously unexplored rationale for specific targeting of PP2A in SCLC cells. LB-100 is a potent and selective antagonist of PP2A that has shown efficacy in a number of pre-clinical models. LB-100 has shown in vitro and in vivo activity as a single agent as well as potentiating the activity of cytotoxic agents including temozolomide (TMZ), doxorubicin (DOX), docetaxel and ionizing radiation in vivo (14). LB-100 is active in combination with TMZ or DOX against xenografts of glioblastoma multiforme (GBM) and neuroblastoma (16, 23), pheochromocytoma (24), breast cancer (mouse and human, unpublished), fibrosarcoma, and melanoma (unpublished). Our recent studies have also shown that LB-100 has in vitro and in vivo activity in combination with carboplatin, etoposide against cell lines and xenografts of small cell lung cancer (unpublished). Racemic LB-100 used alone has modest single agent antitumor activity in vivo against diverse cell types of human cancer. Combined with either TMZ or DOX, LB-100 potentiates their single agent activity, leading to regression of human cancer xenografts for periods of time greater than achieved with either of the standard cytotoxic drugs alone. Thus, the clinical potential of LB-100 lies in using it in combination with chemotherapy.
The combination of LB-100 with carboplatin/etoposide/atezolizumab, the standard of care of ED-SCLC, will be evaluated in treatment naïve patients to determine the RP2D of LB-100, the safety of the combination and to collect preliminary data on whether benefit to progression free survival is observed.
2.3 Preclinical Toxicity Studies of LB-100
2.3.1 LB-100 in Rats
In a non-GLP dose ranging study in rats conducted by the NCI, LB-100 was administered by daily intravenous (IV) infusion at 0.5, 0.75 and 1.5 mg/kg for 4 consecutive days. A no-observed-adverse-effect-level (NOAEL) was not established in this study. The MTD was 0.75 mg/kg (about 4.5 mg/m2) when administered IV daily for 4 days. At 1.5 mg/kg/day, clinical observations included blood in urine (Day 4), lethargy (Day 3 and 4), and hind limb paresis (Day 4). At 1.5 mg/kg/day, adverse effects in kidney (nephrosis) in the distal convoluted tubules were seen in 3 of 3 rats; in the 0.75 mg/kg/day group, nephrosis was mild, and in the 0.5 mg/kg/day group, nephrosis was minimal. Primary clinical signs of blood in the urine and clinical chemistry findings of increased blood urea nitrogen and creatinine supported kidney and urinary bladder as target organs of toxicity. The transient hind limb paresis observed at 1.5 mg/kg/day dose level had no histopathology correlates that would explain the paresis. Heart toxicity (epicardial hyperplasia with inflammation primarily on the epicardium of the atria) was observed in the 0.75 and 1.5 mg/kg/day groups. The hyperplasia was accompanied by subepicardial accumulation of mononuclear cells and eosinophils. One rat in the 1.5 mg/kg/day group had a large focus of inflammation with eosinophils associated with the aorta. Kidney, heart, femoral bone, liver and urinary bladder toxicity appeared to be dose-limiting toxicities in rats treated with LB-100 when administered IV once per day for 4 consecutive days.
| Page 17 of 92 |
In the GLP repeat-dose study in rats, LB-100 administered via daily intravenous (slow bolus) injection for 5 consecutive days to male and female Sprague Dawley rats at dose levels of 0.5, 0.75, and 1.25 mg/kg/day resulted in adverse, test article-related nephrosis of the kidneys in the 0.75 and 1.25 mg/kg/day group males and females, which persisted or progressed in the 0.75 and 1.25 mg/kg/day group males at the recovery necropsy. Test article-related effects on urinalysis parameters were observed in all treatment groups and included an increased in incidence and severity of urine occult blood in 0.5 mg/kg/day group males and 0.75 and 1.25 mg/kg/day group males and females, urine protein in 1.25 mg/kg/day females, and increase in microscopic observations of leukocytes in males and females of the 1.25 mg/kg/day group, and in one female in both the 0.5 and 0.75 mg/kg/day group on Day 5. These changes were reversible. LB-100 administration resulted in subacute subepicardial inflammation and/or mesothelial hypertrophy in the atria of males at ≤ 0.5 mg/kg/day and at 1.25 mg/kg/day in the females at the primary necropsy and was considered adverse in one 1.25 mg/kg/day group male. Minimal to mild subacute inflammation was observed in the epicardium and subepicardium of the left and/or right atrium of the heart in the 0.5, 0.75, and 1.25 mg/kg/day group males and the 1.25 mg/kg/day group females. One male in the 1.25 mg/kg/day group had mild subacute inflammation that was accompanied by minimal fibroplasia (plump fibroblasts) in the right atrium. Inflammation was often accompanied by mesothelial hypertrophy. There was a higher incidence of mesothelial hypertrophy in the 1.25 mg/kg/day group females when compared to the control group. Based on these findings, the severely toxic dose in 10% of the animals (STD 10) for this study was determined as 0.75 mg/kg/day. This dose corresponded to AUClast values of 596 and 691 ng*h/ml and C0 values of 1804 and 2347 ng/ml for males and females, respectively, on study Day 4.
2.3.2 LB-100 in Dogs
In a non-GLP dose ranging study, LB-100 administered intravenously (slow bolus push) to beagle dogs at dose levels of 0.1, 0.25, 0.5, and 1.0 mg/kg given every 4 days x 4 doses (on study days 0, 4, 8, and 12) resulted in a no-observed-effect level (NOEL) of 0.25 mg/kg. There were no LB-100-related effects on survival. A possible test article-related clinical observation of intermittent tremors was noted in one female on study Day 13 following administration of LB-100 at 1.0 mg/kg. At dose levels of 0.5 and 1.0 mg/kg, lower body weight gains and food consumption were noted in females.
In the GLP repeat dose dog study, LB-100 was administered by intravenous injection (slow bolus push) at dose levels of 0.15, 0.30, and 0.75 mg/kg daily for 5 consecutive days. Test article-related lethality was observed 2 of 10 animals in the 0.75 mg/kg/day group, a male and a female were found dead prior to administration of the fourth scheduled dose. The dosage level was reduced to 0.50 mg/kg/day for the 4th and 5th doses (study days 3 and 4). Both animals dying after the 3rd dose at 0.75mg/kg had similar test article-related macroscopic and microscopic findings affecting the gastrointestinal tract, kidneys, injection sites, spleen, larynx, lungs (including acute inflammation) and/or liver. Both animals had experienced emesis, decreased defecation, yellow and red mucoid feces, and red diarrhea; these changes were also observed in animals treated at the 0.3 mg/kg/day dose level.
Although the most noteworthy findings (mitotic figures and single cell necrosis of the renal tubular epithelial cells from the outer medulla and cortex) could be associated with altered renal function, these findings were not considered fatal lesions; therefore, the cause of death for each animal was considered undetermined but directly attributed to test article administration. Note: a dose of 0.75 mg/kg in the dog (average weight of 9 kg and BSA of 0.5 m2) is about 13.8 mg/ m2 or more than twice the MTD in the rat. This highest dose was selected because the dose range study in the dog revealed almost no signs of toxicity following a single dose of 1.0 mg/kg (approximately 18 mg/ m2) in the dose ranging study. All other animals survived to the scheduled primary (study day 5) and recovery (study day 29) necropsies including the dogs receiving 0.75 mg/kg daily for 3 days and 0.5mg/kg for doses 4 and 5. Test article-related histological changes at the Day 5 necropsy included erosion and focal hemorrhage within the gastrointestinal tract in the 0.75/0.5 mg/kg/day dose group. Single cell necrosis was observed throughout the gastrointestinal tract. These changes were reported as resolved in the recovery period. There were no ophthalmic findings or changes in electrocardiography parameters and blood pressures associated with test article administration in any treatment group.
| Page 18 of 92 |
During the recovery period, all surviving animals had body weight gains indicative of recovery, and the majority of the other observed clinical signs resolved within the first few days of the recovery period. At the primary necropsy (Day 5), test article-related macroscopic findings consisted of dark red discoloration of the kidneys, small spleens, and red discoloration (reddened mucosa or dark red area) of various segments of the gastrointestinal tract in the 0.75/0.50 mg/kg/day group males and females. At the recovery necropsy (Day 29), no test article-related macroscopic finding were observed. The primary cause of small spleen size appeared to be due to less blood in the red pulp. Mild or moderate single cell (lymphoid) necrosis was seen in spleens microscopically. Test article-related effects on hematology and coagulation parameters at hematocrit), lower platelet counts, and prolonged activated partial thromboplastin time values in the animals of the 0.75/0.50 mg/kg/day group. In this group, lower platelet counts were statistically significantly lower only in the males, with the group mean level lower than the historical control group mean level. Lower platelet count in a female was not statistically significant but was considered test-article related. At the Day 29 evaluation, there were no residual effects of test article administration on hematology or coagulation parameters. Test-article related changes in urinalysis parameters observed at the day 5 evaluation included lower specific gravity, higher urine volume, and increased presence of blood in the 0.75/0.50 mg/kg/day groups. At day 29, no test article-related changes in urinalysis parameters were present.
Multilead (I, II, III, aVR, aVL, aVF, and V2) ECGs were recorded for all animals prior to randomization (Day -8) and for all surviving animals on Day 4 (recorded approximately 2 to 4 hours following dose administration) and Day 27. All the ECGs were qualitatively and quantitatively interpreted and within normal limits. No test article-related effects attributable to the test article administration were found at any dose level based on comparison of pretest and post-dosing group mean values and control values. No abnormalities in rhythm were found.
Blood pressure (systolic, diastolic, and mean arterial pressure) data were recorded for all animals once during the pretest period (Day -8) and for all surviving animals on study Day 4 (recorded approximately 2 to 4 hours following dose administration) and Day 27. Blood pressure was unaffected by test article administration. There were no statistically significant differences at the Days 4 and 27 evaluations when the control and test article-treated groups were compared.
In conclusion, administration of LB-100 via daily intravenous (slow bolus) injection for 5 consecutive days to male and female beagle dogs was well tolerated at the dosage level of 0.15 mg/kg/day. At dosage levels of 0.30 and 0.75/0.50 mg/kg/day, administration of LB-100 resulted in adverse clinical observations, lower body weights, and histological findings (congestion and nephrosis in kidneys, increased mitoses and single cell necrosis in liver, lymphoid depletion and single cell necrosis in thymus, and/or erosion and/or hemorrhage in stomach or intestines) correlating with effects on clinical pathology, organ weight, and/or macroscopic findings during the dosing period. Persistent adverse test article-related histological changes in the kidneys were observed in the 0.30 and 0.75/0.50 mg/kg/day group males and females at the Day 29 recovery necropsy. These changes were more indicative of a progression towards chronicity rather than recovery. In addition, lethality was observed at 0.75 mg/kg/day. Therefore, the Highest Non-Severely Toxic Dose (HNSTD) was 0.15 mg/kg, which corresponded to an AUClast for LB-100 of 267 and 335 ng•h/mL on study Day 4 for males and females, respectively.
| Page 19 of 92 |
(i) 2.4 Human Studies
2.4.1 Single-Agent Phase I Study LB-100
A single Phase 1 clinical trial with LB-100 in solid tumors (NCT01837667) has been completed (Chung et al. 2016). This open-label, multicenter, dose-escalation, non-randomized, phase I study was performed to assess the safety, tolerability, and activity of intravenous LB-100 administered for 3 consecutive days every 3 weeks. Pharmacokinetic studies were planned on three patients at the maximum tolerable dose (MTD). The starting dose of 0.25mg/m2 intravenously daily for 3 days every 3 weeks was approximately 1/15th of the highest non-severely toxic dose in the dog.
LB-100 for Injection was supplied as a sterile, single use solution. The tested dose levels were 0.25, 0.50, 0.83, 1.25, 1.75, 2.33, and 3.1mg/m2. For doses up to 2.33mg/m2/day, LB-100 was administered in 50mL of saline over 15 minutes. Beginning with the 2nd patient at 2.33mg/m2, LB-100 was administered in 500 mL normal saline over 2 hours because of grade 3 asymptomatic reversible increases in serum creatinine in 2 of 4 patients. The larger fluid volume of 500 mL saline was used to assure adequate hydration.
Patients were eligible to receive up to 6 cycles of study therapy, unless unacceptable toxicity, disease progression, or inter-current illness required discontinuation. Patients were allowed continue beyond 6 cycles if the responsible physician determined that additional treatment might provide benefit. A total of 28 patients with advanced solid tumors were enrolled at four clinical sites. Four patients were not evaluable for toxicity. Three of these had disease-associated complications prior to completing cycle 1: a pulmonary embolism (NSCLC); hypoxia secondary to pulmonary metastases (NSCLC); intra-tumor hemorrhage and chronic anemia (uterine cancer). A fourth patient, with atypical carcinoid of the lung, received one dose of LB-100 but was removed from study before dose 2 because of pneumonia; this patient was re-entered on study 7 weeks later and achieved stable disease for 5 cycles. None of these adverse events was considered related to drug administration. A colon cancer patient had a grade 2 increase in normal pre-treatment creatinine after the second dose of LB-100 at 2.33 mg/m2. The treating physician elected not to administer the 3rd dose although to do so would have been permitted by protocol.
There were 24 patients who completed at least one 3-day cycle of LB-100. There was no symptomatic toxicity other than reversible mild to moderate fatigue. There was no grade 4 or 5 toxicity. There was no DLT during the first 6 dose levels. At the 3.1 mg/m2 dose level, one patient with prostate cancer and one with chondrosarcoma had no DLT during 4 and 9 cycles of treatment, respectively. A third patient with ovarian cancer had a grade 3 increase in calculated creatinine clearance after cycle 1 with a return to normal by day 8 and received 3 more cycles at a reduced dose of 2.33 mg/m2 before tumor progression. A fourth patient, with fibrosarcoma, had a grade 3 increase in calculated creatinine clearance after the first course of LB-100 at 3.1 mg/m2. Because the creatinine returned to pretreatment value by day 21 and the patient was asymptomatic, the safety review committee and sponsor approved a second course at a reduced level of 2.33 mg/m2. This dose level was associated with a grade 2 increase in creatinine clearance without other toxicity. Further treatment at 1.75 mg/m2 was given for ten more cycles without toxicity and persistence of stable disease until disease progression after 36 weeks. Because 2/4 patients at 3.1mg/m2 had grade 3 increases in calculated creatinine clearance during cycle one, three additional patients were evaluated at the preceding dose level of 2.33 mg/m2 and had no limiting toxicity. There were no signs or symptoms suggestive of hepatic, hematologic, neurologic, or immunologic toxicity.
| Page 20 of 92 |
Thus, LB-100 was well tolerated at all dose levels explored during the Phase I study (0.25 mg/m2, 0.5 mg/m2, 0.83 mg/m2, 1.25 mg/m2, 1.75 mg/m2, 2.33 mg/ m2 and 3.1 mg/m2) with few adverse events (AEs) and no drug related serious adverse events (SAEs). During the dose-escalation phase (Arm 1), the maximum tolerated dose (MTD) was identified at 2.33 mg/ m2 daily for 3 days every 3 weeks administered IV. Dose limiting toxicities of Grade 3 increased in creatinine in 2 of 4 patients were observed at 3.1 mg/m2. The most common AEs that were present in more than one patient were fatigue, blood creatinine increase, aspartate aminotransferase increase, headache, hypematremia, hypoalbuminemia, nausea, proteinuria, pyrexia, alanine aminotransferase increase, constipation, neuropathy peripheral, edema peripheral, sinus tachycardia, and anemia.
As LB-100 has not been studied to date in combination with carboplatin/etoposide/atezolizumab, the initial dose level for this study (i.e. 1.75mg/m2 on day 1 and day 3 of the carboplatin/etoposide/atezolizumab regimen) was chosen at one dose level below that which is tolerated in solid cancer patients given for three consecutive days with allotment of dose de-escalation/escalation based on safety and tolerability.
2.4.2 LB-100 Human Pharmacokinetics
Plasma concentrations of LB-100 and endothall were measured at the MTD of 2.33 mg/m2 of LB-100 administered as an intravenous infusion over two hours. In one patient sampling was done on day 1 and in two patients, on day 1 and 3. Plasma samples were obtained prior to the infusion and over 4 hours after completion of the infusion. The pharmacokinetics of LB-100 were characterized by low clearance and a low volume of distribution resulting in a short half-life on day 1. The day 3 pharmacokinetic profile was similar to that described for the first dose. Circulating plasma concentrations of endothall were low over the four-hour infusion. In one patient, concentrations of endothall were below the limit of detection (5 ng/mL) at all time points. For the other two patients, the maximal concentration of endothall was observed at the last sampling timepoint (4 h), which precluded determination of the elimination half-lives for endothall.
2.4.3 LB-100 Anti-tumor Activity in Solid Tumor Patients
Of 20 patients with measurable disease, one patient with pancreatic cancer had a partial regression, noted after 10 cycles and lasting for 5 more cycles, and 16 patients had no progression of their indicator lesion. Only 3 patients, one with duodenal and two with colonic adenocarcinomas, had significant increases in the size of their indicator lesion by RECIST criteria and were removed from study for either the appearance of a new lesion or symptoms judged to represent clinical progression. Achieving partial regression or stability of disease was not clearly dose-dependent, occurring at 0.83 mg/m2 in pancreatic cancer (15 cycles) and atypical carcinoid of the lung (5 cycles); at 1.25 mg/m2 in breast cancer (4 cycles) and testicular cancer (5 cycles); and at 1.75 mg/m2 in malignant thymoma (8 cycles) and ovarian cancer (6 cycles). At 3.1 mg/m2, a patient with chondrosarcoma was stable for 8 cycles of LB-100 without any alteration in normal renal function whereas a patient with fibrosarcoma started at 3.1 mg/m2 was stable for 12 cycles after two dose reductions. Thus ten of 20 patients had stable disease for four or more cycles with one pancreatic adenocarcinoma patient exhibiting partial response after 10 cycles, which was maintained for five additional cycles. It was noted that the stabilization of disease occurred over a range of doses (0.83-2.33 mg/m2 daily for 3 days). The only expected toxicity from LB-100 is reversibly increases in serum creatinine that may result from transient reversible inhibition of PP2A in the renal tubules and not from renal tissue damage. As discussed earlier, we expect LB-100 to increase the mechanisms of antitumor activity of carboplatin and etoposide which could increase their toxicity. However, the effect of LB-100 on the toxicity of the PD-L1 compound is unknown.
| Page 21 of 92 |
2.4.4 LB-100 Dosage and Schedule Considerations
The optimal rate of LB-100 infusion and the dosing schedule are not known. In the Phase 1 trial, as specified per protocol, PK was done was done in three patients at the MTD, 2.33 mg/m2 in 500 mL saline over two hours. Two were sampled day 1 and day 3, and one on day 1 only. Peak plasma concentrations of LB-100 at the end of the infusion (n=5) averaged 150 ng/ml (~ 0.6 uM) and declined rapidly. As expected, at the end of the two-hour infusion in all patients on day 1 infusion, there was no detectable endothall in plasma. In one of two patients, endothall was present at 17.6 ug/mL (~0.1 uM) in the pretreatment sample on day 3 before the 3rd infusion, suggesting that 24 hours after the day 2 infusion, some endothall was still in tissues, compatible with its relatively slow elimination (t ½ ~7 hours) compared to LB-100 with a t 1/2 of less than 1 hour. However, there is no estimate of the concentrations of LB-100 and endothall in tissues and PK studies were not done after infusion of LB-100 over 15 minutes. Based on the above data for this trial and subsequent studies, it is recommended that LB-100 be infused over 15 minutes on days 1 and 3 of the standard carboplatin/etoposide/atezolizumab regimen to maximize peak plasma concentration and to avoid tissue accumulation of the active metabolite, endothall, which is now known to have a plasma half-life of up to 7 hours. Thus, the Investigator Brochure may be updated during the course of this study with additional risks and benefits. Please see the current Investigator Brochure for further details about the potential risks and benefits associated with this study.
In the Phase 1 trial of LB-100 in solid tumor patients, three patients received a dose of 1.75 mg/m2 in 50 mL normal saline over 15 minutes daily for three consecutive days without any significant toxicity. In the present trial the starting dose of LB-100 is 1.75 mg/m2 in 50 mL normal saline on day 1 and day 3. Thus, we anticipate no toxicity attributable to LB-100 itself, allowing recognition of any potentiation of the standard toxicities associated with the other agents.
LB-100 is supplied as a sterile solution for intravenous administration. LB-100 is stored at -20C (range: - 25C to -10C). Each vial contains LB-100 at a concentration of 1 mg/mL. The proper dose is drawn up in a sterile syringe and added to 50 mL of normal saline (0.9%) and infused over 15 +/- 5 minutes. Following dilution in normal saline, LB-100 should be administered within 4 hours.
2.5 Carboplatin/Etoposide/Atezolizumab
Carboplatin is an analog of cisplatin that has a more favorable toxicity profile (Ruckdeschel 1994). In interacts with DNA and forms both intra- interstrand links. The most commonly observed side effects include thrombocytopenia, neutropenia, leukopenia, and anemia. Like other platinum-containing compounds, carboplatin may induce anaphylactic-type reactions such as facial edema, wheezing, tachycardia, and hypotension that may occur within a few minutes of drug administration. These reactions may be controlled with adrenaline, corticosteroids, or antihistamines. Please see package insert for further information.
Etoposide is a semisynthetic derivative of podophyllotoxin that exhibits cytostatic activity in vitro by preventing cells from entering mitosis or by destroying them at a premitotic stage. Etoposide interferes with the synthesis of DNA and appears to arrest human lymphoblastic cells in the late S-G2 phase of the cell cycle. The most commonly observed side effects include leukopenia and thrombocytopenia. Please see package insert for further information.
Etoposide is indicated in combination with other antineoplastics in the treatment of SCLC, NSCLC, malignant lymphoma, and testicular malignancies (approved indications may vary depending on the specific country). Etoposide is also used in clinical studies against many other types of cancer including head and neck, brain, bladder, cervical, and ovarian.
| Page 22 of 92 |
Atezolizumab is a humanized immunoglobulin (Ig) G1 monoclonal antibody that targets programmed death receptor 1 ligand (PD-L1) and inhibits the interaction between PD-L1 and its receptors, programmed death receptor 1 (PD-1) and B7-1 (also known as CD80), both of which function as inhibitory receptors expressed on T cells. Intravenous atezolizumab has been approved in the US and EU for the treatment of adult patients with advanced urothelial carcinoma that have failed or are ineligible for a platinum based regimen.(25, 26) Additionally, atezolizumab in combination with bevacizumab, paclitaxel, and carboplatin has been approved in the US for the first-line treatment of adult patients with metastatic NSCLC with no EGFR or ALK genomic tumor aberrations and as monotherapy in locally advanced and metastatic NSCLC after prior chemotherapy.(27) Recently, atezolizumab was also granted accelerated approval in the US, in combination with nab-paclitaxel for patients with unresectable locally advanced or metastatic triple negative breast cancer whose tumors express PD-L1.(28) Finally, atezolizumab was approved for first-line treatment, in combination with carboplatin and etoposide, in adult patients with extensive-stage small cell lung cancer, showing improved survival (median OS 12.3 months in the platinum/etoposide/atezolizumab arm vs. 10.3 months platinum/etoposide/placebo). The addition of immunotherapy to etoposide and platinum chemotherapy in ED-SCLC also improved progression-free survival and was not associated with unacceptable toxicity. (7) Treatment with atezolizumab is generally well-tolerated, but can be associated with immune-related adverse events (irAEs). Please see package insert for further information.
| 3.0 | Patient Eligibility | |
| 3.1 | Inclusion Criteria |
Patients are eligible to be included in the study only if they meet all of the following criteria:
3.1.1 Disease Status
| [1] | histologically or cytologically confirmed extensive-stage disease small cell lung carcinoma per the Veterans Administration Lung Study Group (VALG) staging system, (Appendix E) |
| [2] | measurable disease as defined by Response Evaluation Criteria in Solid Tumors (RECIST): Revised RECIST Guideline (version 1.1; Eisenhauer et al. 2009, Appendix B) |
3.1.2 Age Criteria, Performance Status, and Life Expectancy
| [3] | at least 18 years old at the time of screening |
| [4] | estimated life expectancy of at least 12 weeks |
3.1.3 Childbearing Potential
| [5] | For women: Must be surgically sterile (surgical procedure: bilateral tubal ligation), post-menopausal (at least 12 consecutive months of amenorrhea) or have a negative pregnancy test. Women of childbearing potential must be compliant with a medically approved contraceptive regimen (intrauterine device [IUD], birth control pills, or barrier device) during and for 3 months after the treatment period; must have a negative serum or urine pregnancy test within 14 days before study drug treatment and must not be breastfeeding. |
For men: agreement to remain abstinent or use medically approved contraceptive measures, as defined below:
With female partners of childbearing potential or pregnant female partners, men must remain abstinent or use a condom during study therapy and for at least 6 months after the last dose of study therapy to avoid exposing the embryo.
| Page 23 of 92 |
3.1.4 Protocol-Specific Criteria
| [6] | a performance status of 0-2 on the Eastern Cooperative Oncology Group (ECOG) scale (Appendix A) |
| [7] | adequate hematologic and organ function, including: |
Hematologic: absolute neutrophil (segmented and bands) count (ANC) ≥1.5x10/L, platelets ≥100x10/L, and hemoglobin ≥9 g/dL
Hepatic: bilirubin ≤1.5 times upper limits of normal (ULN) may be enrolled, and alkaline phosphatase (AP), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) ≤3.0 times ULN (AP, AST, and ALT ≤5 times ULN are acceptable if the liver has tumor involvement).
Renal: calculated creatinine clearance (CrCl) ≥60 mL/min based on the Cockcroft and Gault formula below:
| Males: | ||||
| Creatinine CL | = | Weight (kg) x (140 – Age) . | ||
| (mL/min) | 72 x serum creatinine (mg/dL) | |||
| Females: | ||||
| Creatinine CL | = | Weight (kg) x (140 – Age) | x 0.85 | |
| (mL/min) | 72 x serum creatinine (mg/dL) |
3.1.5 Informed Consent/Assent
| [8] | All subjects must have the ability to understand and the willingness to sign a written informed consent. |
3.1.6 Prior Therapy
| [9] | no prior systemic chemotherapy, immunotherapy, biological, hormonal, or investigational therapy for SCLC |
3.2 Exclusion Criteria
| [10] | currently enrolled in, or discontinued within the last 30 days from, a clinical trial involving an investigational product or non-approved use of a drug or device, or concurrently enrolled in any other type of medical research judged not to be scientifically or medically compatible with this study |
| [11] | diagnosis of NSCLC or mixed NSCLC and SCLC |
| [12] | no prior malignancy other than SCLC, carcinoma in situ of the cervix, or nonmelanoma skin cancer, unless that prior malignancy was diagnosed and definitively treated 5 or more years prior to study entry with no subsequent evidence of recurrence. Patients with a history of low grade (Gleason score ≤6=Gleason Group 1) localized prostate cancer will be eligible even if diagnosed less than 5 years prior to study entry |
| [13] | serious concomitant systemic disorder that, in the opinion of the investigator, would compromise the patient’s ability to adhere to the protocol |
| [14] | active or ongoing infection during screening requiring the use of systemic antibiotics |
| Page 24 of 92 |
| [15] | serious cardiac condition, such as myocardial infarction within 6 months, angina, or heart disease as defined by the New York Heart Association Class III or IV |
| [16] | clinical evidence of central nervous system (CNS) metastases or leptomeningeal carcinomatosis, except for individuals who have previously-treated CNS metastases, are asymptomatic, and have had no requirement for steroid medication for 1 week prior to the first dose of study drug and have completed radiation 2 weeks prior to the first dose of study drug |
| [17] | known or suspected allergy to any agent given in association with this trial |
| [18] | pregnant or lactating women |
| [19] | History of autoimmune disease, including minor/mild autoimmune disease not requiring immunosuppressants (such as eczema on less than 10% of the body surface area and long term diabetes mellitus type 1on stable insulin). |
| [20] | Known hepatitis B or hepatitis C |
| [21] | Known human immunodeficiency virus (HIV) positive |
| [22] | Treatment with systemic corticosteroid or other immunosuppressive medication. The use of inhaled corticosteroids for chronic obstructive pulmonary disease, mineralocorticoids (e.g., fludrocortisone) for patients with orthostatic hypotension, and low-dose supplemental corticosteroids for adrenocortical insufficiency are allowed. |
| [23] | Administration of a live, attenuated vaccine within 28 days prior to study |
| [24] | Uncontrolled pleural effusion, pericardial effusion, or ascites requiring recurrent drainage procedures (once monthly or more frequently). Patients with indwelling catheters are allowed. |
| [25] | Uncontrolled or symptomatic hypercalcemia (> 1.5 mmol/L ionized calcium or calcium > 12 mg/dL or corrected serum calcium > ULN). Patients who are receiving denosumab prior to study entry must be willing and eligible to discontinue its use and replace it with a bisphosphonate while in the study. |
| [29] | History of idiopathic pulmonary fibrosis, organizing pneumonia (e.g., bronchiolitis obliterans), drug-induced pneumonitis, idiopathic pneumonitis, or evidence of active pneumonitis on screening chest CT scan. History of radiation pneumonitis in the radiation field (fibrosis) is permitted. |
| [30] | Prior allogeneic bone marrow transplantation or solid organ transplant. |
| [31] | QTcF (Fridericia Correction Formula) > 470 on 2 out of 3 EKG’s. |
| [32] | Diagnosis of congenital long QT syndrome. |
| [33] | Treatment, within 7 days prior to first dose of study drug, with medications that are known to prolong the QT interval and/or are associated with a risk of Torsades de Pointes. |
| [34] | Treatment with CYP450 substrates (see Appendix F) within 7 days prior to first dose of study drug. |
| [35] | Treatment with nephrotoxic compounds within 7 days prior to first dose of study drug. |
| [36] | Treatment with warfarin within 7 days prior to first dose of study drug. |
| [37] | Treatment with antiepileptic medications that may increase etoposide clearance (including but not limited to phenytoin, phenobarbital, carbamazepine, and valproic acid) within 7 days prior to first dose of study drug. |
| [38] | Treatment with strong P-glycoprotein inhibitors within 7 days prior to first dose of study drug. |
3.2.1 Non-Compliance
| [39] | Subjects, who in the opinion of the investigator, may not be able to comply with the safety monitoring requirements of the study. |
| Page 25 of 92 |
3.3 Inclusion of Women and Minorities
The study is open to anyone regardless of gender or ethnicity. Efforts will be made to extend the accrual to a representative population, but in a trial which will accrue approximately 18 subjects, a balance must be struck between subject safety considerations and limitations on the number of individuals exposed to potentially toxic or ineffective treatments on the one hand and the need to explore gender, racial, and ethnic aspects of clinical research on the other. If differences in outcome that correlate to gender, racial, or ethnic identity are noted, future studies may explore those differences.
| 4.0 | Screening and Registration Procedures | |
| 4.1 | Pre-Enrollment Informed Consent and Screening Procedures |
Diagnostic or laboratory studies performed exclusively to determine eligibility will be done only after obtaining written informed consent. Studies or procedures that are performed for clinical indications (not exclusively to determine study eligibility) may be used for baseline values and/or to determine pre- eligibility, even if the studies were done before informed consent was obtained.
The informed consent process is to be fully documented (see Section 14.7), and the prospective participant must receive a copy of the signed informed consent document. Screening procedures are listed in Section 10.0 (Study Calendar).
4.2 Participant Enrollment
4.2.1 COH DCC Availability and Contact Information
Eligible participants will be registered on the study centrally by the Data Coordinating Center (DCC) at City of Hope.
DCC staff are available between the hours of 8.00 am and 5.00 pm PST, Monday through Friday (except holidays).
| o | E-mail: DCC@coh.org |
4.2.2 Slot verification and reservation
A designated study team member should email the DCC to verify current slot availability, and to reserve a slot for a specific prospective subject (provide DCC with subject initials), including a tentative treatment date. Slots can only be held for a limited time, at the discretion of the study PI.
The DCC should be notified of cancellations of prospective participants holding slots as soon as possible.
4.2.3 Registration Process
Allow up to 24 hours for the DCC to review eligibility. To register a participant the subsequent procedure is to be followed:
| 1. | The study team should contact the DCC via email to provide notification regarding the pending registration and communicate desired timeline of the registration, especially if it must be completed promptly to meet the registration window. |
| Page 26 of 92 |
| 2. | The study team will email a Complete Eligibility Packet to the DCC, which consists of a copy of the following documents: |
| o | Registration Cover Sheet (Appendix C) | |
| o | Completed eligibility checklist (printed from Section 3.0 of the protocol) with required signature(s) | |
| o | Source documents that support all eligibility criteria listed in the eligibility checklist | |
| o | Signed Informed Consent | |
| o | Signed HIPAA authorization form (if separate from informed consent) | |
| o | Signed subject’s bill of Rights (California only) |
| 3. | When all source documents are received, the DCC will review to verify eligibility, working with the study team to resolve any missing required source elements. Any missing documents may delay review and registration. A participant failing to meet all protocol eligibility requirements will not be registered and the study team will be immediately notified. |
| 4. | Once eligibility is confirmed, the DCC will send a Confirmation of Registration Form and signed Eligibility Checklist, including the Subject Study Number and cohort assignment to: |
| o | The study team: Site Lead Investigator, treating physician/sub-investigator, protocol nurse, CRC and pharmacy (as needed). | |
| o | The COH Study PI and COH study team designees (including but not limited to study monitor(s) and statistician(s)). |
| 5. | Upon receipt of the Confirmation of Registration Form, COH study team will register the patient in OnCore. |
4.3 Screen Failures and Registered Participants Who Do Not begin Study Treatment
Notify the DCC immediately if the participant screen fails after registration or if the participant does not start treatment. Issues that would cause treatment delays should be discussed with the Study Principal Investigator.
4.4 Screening Procedures
Diagnostic or laboratory studies performed exclusively to determine eligibility for this trial will be done only after obtaining written informed consent. Studies or procedures that were for clinical indications (not exclusively to determine study eligibility) may be used for baseline values, even if the studies were done before informed consent was obtained. Reference is made to Section 10.0 – Study Calendar.
4.5 Informed Consent
The investigational nature and objectives of the trial, the procedures and treatments involved and their attendant risks and discomforts, and potential alternative therapies will be carefully explained to the subject and a signed informed consent will be obtained. Documentation of informed consent for screening will be maintained in the subject’s research chart and medical record.
4.6 Registration Requirements/Process
Prior to registration and any study-specific evaluations being performed, all patients must have given written informed consent for the study and must have completed the pre-study evaluations (see Section 10.0). Patients must meet all of the eligibility requirements listed in Section 3.0. Patients will be registered on the study by DCC, after review of the eligibility criteria checklist, source documentation, informed consent, HIPAA and Bill of rights.
| Page 27 of 92 |
4.7 Dose Level Assignment
Prior to enrollment into the study, an eligibility check must be conducted for every patient by the investigational site to confirm that the patient meets all enrollment criteria.
| 5.0 | Treatment Program | |
| 5.1 | Treatment Overview |
This Phase Ib study of LB-100 diluted in 50 mL of normal saline for injection will be administered intravenously in the outpatient clinic over 15 minutes in patients with extensive-stage small cell lung cancer.
5.1.1 Schedule
For a tabular view of the treatment monitoring and follow-up schedule, see study calendar in Section 10.
Patients will receive an intravenous infusion of LB-100 diluted in 50 mL of normal saline (0.9%) over 15 +/- 5 minutes on days 1 and 3 of each 21 day cycle at escalating doses starting at Dose Level 1 (see Table 5.1). The LB-100 should be given first and should end one hour before the start of other drugs. All three patients at each dose level will be assessed for evidence of limiting toxicity through their return visit day 21 (and any delay prior to the start of cycle 2) before the decision is made for dose escalation in the next cohort. The MTD is defined as the highest dose level below which DLT is manifested in ≥33% of the patients (unless the highest dose to be tested does not have ≥33% of patients with a DLT) and where at least 6 patients have been treated.
The study is based on a standard 3+3 patient dose escalation design. It is planned that there will be 3 possible dose escalations (and one possible de-escalation level if needed). Thus, a maximum of 24 patients will be enrolled during dose finding, with an expected sample-size of 12 during escalation/de-escalation (additional patients to achieve 12 patients at the RP2D will follow for an expected sample-size of 18 total patients and maximum of 30).
All patients who are not evaluable for DLT (dose-limiting toxicity) will be replaced. Patients who do not receive the planned doses without a DLT, will be considered inevaluable as will patients where inadequate follow-up assessments are conducted for reasons unrelated to toxicity. Patients will be enrolled at most in cohorts of 3.
If 0/3 patients have a DLT attributable to the combination, then the next 3 patients will be treated at the next dose level.
If a DLT treatment occurs in 1/3 patients, then 3 more patients (for a total of 6) will be treated at the same dose level. If no additional DLT attributable to treatment is observed at the expanded dose level (i.e. 1/6 with DLT), then the LB-100 dose will be escalated to the next level. If two or more patients (i.e. 2/6) have a DLT then one level below that dose will be tested.
Dose escalation will terminate as soon as two or more patients have a DLT at a given dose level or the highest dose level is tested.
There will be no dose escalation within a patient.
The MTD is defined as the highest LB-100 dose tested in which none or only one patient had a DLT during the first cycle of therapy, when at least six patients were treated at that dose and are evaluable for toxicity assessment. The MTD is one dose level below the lowest dose tested in which 2 patients had a DLT attributable to treatment unless the highest dose is deemed safe.
| Page 28 of 92 |
In addition to these rules, all dose modifications and later cycle toxicities will be reviewed prior to escalation or expansion and can modify the decision to be more conservative (e.g. to not escalate when the standard rules state escalate, or de-escalate when the standard rules state expand the dose).
Any severe immune-related event that requires discontinuation of therapy will also prompt a review by the DSMC, regardless of cycle of therapy.
Table 5.1.1 Dose Levels: LB-100 on Days 1 and 3 of a 21 Day cycle, at escalating doses prior to standard doses of carboplatin/atezolizumab/etoposide
| Dose Level | LB-100 (mg/m2) | |
| -2(a) | 0.50 | |
| -1(b) | 0.83 | |
| 1 (Starting dose) | 1.25 | |
| 2 | 1.75 | |
| 3 | 2.33 | |
| 4 | 3.10 |
| a) | For within patient dose de-escalation only. | |
| b) | In the event that 2 or more DLT’s are observed at Dose Level 1, subsequent patients will be enrolled in Dose Level -1. |
LB-100:
LB-100 is supplied as a sterile solution for intravenous administration. LB-100 is stored at -20C (range: - 25C to -10C). Each vial contains 10 mL of LB-100 at a concentration of 1 mg/mL. The proper dose is drawn up in a sterile syringe and added to 50 mL of normal saline (0.9%) and infused over 15 +/- 5 minutes prior to administration of atezolizumab on Day 1 and prior to etoposide on Day 3. Following dilution in normal saline, LB-100 should be administered within 4 hours
Carboplatin:
Carboplatin is supplied as a sterile lyophilized powder available in single-dose vials containing 50 mg, 150 mg and 450 mg of carboplatin for administration by intravenous injection. Each vial contains equal parts by weight of carboplatin and mannitol. Immediately before use, the content of each vial must be reconstituted with either Sterile Water for Injection, USP, 5% Dextrose in Water, or 0.9% Sodium Chloride Injection, USP, according to the following schedule:
| Vial Strength | Diluent Volume | |
| 50 mg | 5 mL | |
| 150 mg | 15 mL | |
| 450 mg | 45 mL |
| Page 29 of 92 |
These dilutions all produce a carboplatin concentration of 10 mg/mL. Carboplatin can be further diluted to concentrations as low as 0.5 mg/mL with 5% Dextrose in Water or 0.9% Sodium Chloride Injection, USP (NS).
VP-16 (Etoposide):
100 mg of VP-16 is supplied as 5 mL of solution in Sterile Multiple Dose Vials for injection. The pH of the yellow clear solution is 3-4. Each mL contains 20 mg VP-16, 2 mg citric acid, 30 mg benzyl alcohol, 80 mg polysorbate 80/tween 80, 650 mg polyethylene glycol 300 and 30.5% (v/v) alcohol. VP-16 must be diluted prior to use with either 5% Dextrose Injection, USP or 0.9% sodium Chloride Injection, USP. The time before precipitation occurs depends on concentration, however, when at a concentration of 0.2 mg/mL it is stable for 96 hours at room temperature and at 0.4 mg/mL it is stable for 48 hours.
Atezolizumab (Tecentriq):
Atezolizumab is a sterile, preservative-free, and colorless to slightly yellow solution for intravenous infusion supplied as a carton containing one 1200 mg/20 mL single-dose vial (NDC 50242-917-01). Store vials under refrigeration at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not freeze. Do not shake.
Table 5.1.2 Study drug schedule, dose, route and timing
The induction phase is four cycles (Cycles 1-4). The maintenance phase is Cycle 5 and beyond.
| Page 30 of 92 |
| Drug | Dose | Route | Schedule | Notes | ||||
|
LB-100 (Induction and Maintenance) |
As assigned (.83, 1.25, 1.75, 2.33 or 3.10 mg/m2) |
IV | Days 1 and 3 of each 21 day cycle during the induction phase (Cycles 1-4) and maintenance phase (Cycle 5 onward) | Infused over 15 minutes. Given first. Other drugs should start 1 hour after end of LB-100 infusion. | ||||
|
Atezolizumab (Tecentriq) (Induction and Maintenance) |
1200 mg/20 mL | IV | Day 1 of each 21 day cycle during the induction phase (Cycles 1-4) and maintenance phase (Cycle 5 onward) | Infused over 60 (± 15) minutes (for first infusion, shortening to 30 [± 10] minutes for subsequent infusions, depending on patient tolerance. | ||||
| Carboplatin (Induction) | AUC 5 | IV | Day 1 of the 21 day cycle; repeat every 21 days for 4 cycles | Given after atezolilzumab. Infused over 30-60 minutes. | ||||
| VP-16 (Etoposide) (Induction) | 100 mg/m2 | IV | Days 1, 2 and 3 of the 21 day cycle; repeat every 21 days for 4 cycles | Given last. Infused over 60 minutes. |
5.2 Planned Duration of Therapy
5.2.1 Baseline and Study Treatment Periods
Within 4 weeks before the first dose of study treatment, baseline tumor measurement(s) will be performed on each patient. At baseline: computed tomography (CT) [or magnetic resonance imaging (MRI)] of the head, chest, abdomen, pelvis, and a bone and/or PET scan. Ultrasound will not be permitted as a method of tumor measurement. The same method used at baseline must be used consistently for tumor assessment and will be repeated every 6-8 weeks until disease progression. Confirmation of response will occur no less than 4 weeks from the first evidence of response. A bone and/or PET scan can be repeated per the investigator’s discretion but must be repeated to confirm a complete response (CR) if bone lesions were present at baseline.
Patients may continue to receive study therapy unless unacceptable toxicity, disease progression, intercurrent illness or one of the criteria listed in 5.3 require discontinuation
For reasonable cause, either the Investigator or the Sponsor may terminate this study permanently. Written notification of the termination is required. Conditions that may warrant termination include, but are not limited to.
| ● | The discovery of an unexpected significant or unacceptable risk to the patients enrolled in the study. | |
| ● | Failure of the Investigator to enter patients at an acceptable rate. |
| Page 31 of 92 |
| ● | Insufficient adherence to protocol requirements (non-compliance). | |
| ● | Lack of evaluable and/or complete data. | |
| ● | Decision to modify the developmental plan of the drug. | |
| ● | A decision on the part of the Sponsor to suspend or discontinue development of the drug. |
In the case that the trial is discontinued due to reasons other than unforeseen risk, patients who are currently receiving drug and are deriving benefit from the treatment may be allowed to continue receiving treatment.
5.2.2 Post discontinuation Period
Each enrolled patient will have a 30-day safety follow-up period which will occur 30 days after the last dose of study drug. The investigative sites will continue to monitor patients per routine clinical practice. Patients who complete treatment or discontinue without disease progression will continue to be evaluated for tumor response using the RECIST v1.1 guidelines (Eisenhauer et al. 2009, Appendix B) every 6-8 weeks until disease progression, death, or until study closure, whichever occurs first. The date of first documented disease progression must be recorded on the CRF even if progression occurs after the patient has started a new therapy. Monitoring for survival may also continue following progression on a monthly basis. Information will be collected regarding dates of disease progression, death and any post discontinuation systemic therapy, radiotherapy, or surgical intervention until the date of study closure.
5.3 Criteria for Removal from Treatment
The criteria for enrollment must be followed explicitly. If a patient who does not meet enrollment criteria is inadvertently enrolled, Lixte Biotechnology Holdings, Inc must be contacted. In these cases, the investigator must obtain documented approval John Kovach, M.D. President and CEO of Lixte Biotechnology Holdings, Inc to allow the patient to continue to receive the study drug. Dr. Kovach’s contact information is: telephone 631-880-2907 and email jkovach@lixte.com.
In addition, patients will be discontinued from the study drug and from the study in the following circumstances:
| ● | Enrollment in any other clinical trial involving an investigational product or enrollment in any other type of medical research judged not to be scientifically or medically compatible with this study. | |
| ● | Investigator/Physician Decision |
| o | The investigator/physician decides that the patient should be withdraw from the study or study drug. | |
| o | If the patient, for any reason, requires treatment with another therapeutic agent that has been demonstrated to be effective for treatment of the study indication, discontinuation from the study drug occurs prior to introduction of the new agent. |
| ● | Patient Decision |
| o | The patient [or patient’s designee (for example, parents or legal guardian)] requests to be withdrawn from the study or study drug. |
| ● | Sponsor Decision |
| Page 32 of 92 |
| o | The investigator or DSMB or Sponsor stops the study or stops the patient’s participation in the study for medical, safety, regulatory, or other reasons consistent with applicable laws, regulations, and good clinical practice. |
| ● | The patient is significantly noncompliant with study procedures and/or treatment | |
| ● | The patient has evidence of disease progression | |
| ● | Unacceptable toxicity | |
| ● | The patient becomes pregnant or fails to use adequate birth control (for those patients who are of childbearing potential). |
The reason and date for discontinuation will be collected for all patients. All randomized patients who discontinue regardless of whether they received study drug or not, will have procedures performed as shown in the Study Schedule (Section 10.0).
5.4 Subject Follow-Up
The short-term safety follow-up period begins one day after the last dose of study drug and lasts 30 days. All AEs should be reported for a minimum of 30 days from the last dose of study drug.
The long-term follow-up period begins after patients have either completed cycle 4 or have been discontinued from study drug and continues until disease progression or death. Patients may continue to be followed for survival following progression.
The study will be considered complete following the data cutoff date and data lock for the final analysis. The statistical analysis will be performed after study completion.
5.5 Supportive Care, Other Concomitant Therapy, Prohibited Medications
Premedication with antihistamines may be administered. GCSF may be given in any cycle other than Cycle 1.
The following therapies should continue while patients are in the study:
| ● | Oral contraceptives |
| ● | Hormone-replacement therapy |
| ● | Prophylactic or therapeutic anticoagulation therapy (such as low molecular weight heparin or warfarin at a stable dose level) |
| ● | Palliative radiotherapy (e.g., treatment of known bony metastases) provided it does not interfere with the assessment of tumor target lesions (e.g., the lesion being irradiated is not the only site of disease, as that would render the patient not evaluable for response by tumor assessments according to RECIST v1.1) |
| ● | Inactive influenza vaccinations |
| ● | Megestrol administered as an appetite stimulant |
| ● | Inhaled corticosteroids for chronic obstructive pulmonary disease |
| ● | Mineralocorticoids (e.g., fludrocortisone) |
| ● | Low-dose corticosteroids for patients with orthostatic hypotension or adrenocortical insufficiency |
| Page 33 of 92 |
In general, investigators should manage a patient’s care with supportive therapies as clinically indicated per local standards. Patients who experience infusion-associated symptoms may be treated symptomatically with acetaminophen, ibuprofen, diphenhydramine, and/or famotidine or another H2 receptor antagonist per standard practice. Serious infusion-associated events manifested by dyspnea, hypotension, wheezing, bronchospasm, tachycardia, reduced oxygen saturation, or respiratory distress should be managed with supportive therapies as clinically indicated (e.g., supplemental oxygen and β2- adrenergic agonists).
Cautionary: Systemic corticosteroids and TNF-α inhibitors may attenuate potential beneficial immunologic effects of treatment with atezolizumab. Therefore, in situations where systemic corticosteroids or TNF-α inhibitors would be routinely administered, alternatives, including antihistamines, should be considered first by the treating physician. If the alternatives are not clinically appropriate, systemic corticosteroids and TNF-α inhibitors may be administered per Section 6.3 or after discussion with the Principal Investigator, except in the case of patients for whom CT scans with contrast are contraindicated (i.e., patients with contrast allergy or impaired renal clearance).
Systemic corticosteroids are recommended, with caution, at the discretion of the treating physician, for the treatment of specific adverse events when associated with atezolizumab therapy, per Section 6.3.
Prohibited: Any concomitant therapy intended for the treatment of cancer, whether health authority−approved or experimental, is prohibited for various time periods prior to starting study treatment, and during study treatment until disease progression is documented and patient has discontinued study treatment. This includes, but is not limited to, chemotherapy, hormonal therapy, immunotherapy, radiotherapy, investigational agents, or herbal therapy (unless otherwise noted).
The following medications are prohibited while on study, unless otherwise noted:
| ● | Traditional herbal medicines, because their use may result in unanticipated drug-drug interactions that may cause or confound assessment of toxicity | |
| ● | Denosumab; patients who are receiving denosumab prior to enrollment must be willing and eligible to receive a bisphosphonate instead while in the study | |
| ● | Any live, attenuated vaccine (e.g., FluMist®) within 28 days prior to first study drug, during treatment, or within 90 days following the last dose of atezolizumab | |
| ● | Use of steroids to premedicate patients for whom CT scans with contrast are contraindicated (i.e., patients with contrast allergy or impaired renal clearance); in such patients, non-contrast CT scans of the chest and non-contrast CT scans or MRIs of the abdomen and pelvis should be performed | |
| ● | Medications that are known to prolong the QT interval and/or are associated with a risk of Torsades de Pointes. | |
| ● | CYP450 substrates (see Appendix F). | |
| ● | Nephrotoxic compounds. | |
| ● | Warfarin. | |
| ● | Antiepileptic medications that may increase etoposide clearance (including but not limited to phenytoin, phenobarbital, carbamazepine, and valproic acid). | |
| ● | Strong P-glycoprotein inhibitors. |
5.6 Additional Studies
Not applicable.
| Page 34 of 92 |
5.7 Definition of Dose-Limiting Toxicity (DLT)
The NCI Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0 will be used to grade toxicity. Per section 5.5 GCSF is not allowed in Cycle 1, as it may suppress a toxicity that might otherwise occur. If a protocol deviation occurs and a patient does receive GCSF in Cycle 1, they will be considered inevaluable for DLT and replaced, unless they experience a DLT in Cycle 1. DLT is defined as any of the following adverse events occurring in the first cycle of treatment and considered to be possibly, probably, or definitely related to study treatment:
| ● | Nausea/vomiting of Grade 3 or greater despite maximal antiemetic therapy. | |
| ● | Any Grade 4 (immune-related adverse events (irAE) | |
| ● | Diarrhea of Grade 3 or greater despite maximal antidiarrheal therapy. | |
| ● | Any ≥ Grade 3 colitis (infectious etiologies should have been ruled out and endoscopic verification is strongly encouraged) | |
| ● | Any Grade 3 or 4 noninfectious pneumonitis irrespective of duration | |
| ● | Any Grade 2 pneumonitis that does not resolve to ≤ Grade 1 within 3 days of the initiation of maximal supportive care | |
| ● | Any Grade 3 irAE, excluding colitis or pneumonitis, that does not downgrade to Grade 2 within 3 days after onset of the event despite optimal medical management including systemic corticosteroids or does not downgrade to ≤ Grade 1 or baseline within 14 days | |
| ● | Concurrent elevation of AST or ALT > 3X ULN AND total bilirubin > 2X ULN | |
| ● | AST or ALT > 8X ULN or total bilirubin > 3X ULN, even if asymptomatic, unless it is related to a definite progression of liver metastases or another clearly identifiable etiology. | |
| ● | Grade 4 neutropenia observed for greater than 5 days duration or Grade 3 neutropenia associated with fever of any duration or where sepsis results or Grade 3 neutropenia lasting > 7 days. | |
| ● | Grade 4 thrombocytopenia or Grade 3 thrombocytopenia with clinically significant bleeding or Grade 3 thrombocytopenia lasting > 7 days. | |
| ● | Grade 4 anemia. | |
| ● | Any ≥ Grade 3 AE, except for the exclusions listed below: |
| o | Grade 3 fatigue lasting ≤ 7 days | |
| o | Grade 3 laboratory abnormalities, other than ALT or AST, that are not considered clinically significant and that return to grade 2 or less within 72 hours | |
| o | Grade 3 endocrine disorder (thyroid, pituitary, and/or adrenal insufficiency) that is managed with or without systemic corticosteroid therapy and/or hormone replacement therapy and the subject is asymptomatic | |
| o | Grade 3 inflammatory reaction attributed to a local antitumor response (eg, inflammatory reaction at sites of metastatic disease, lymph nodes, etc.) | |
| o | Concurrent vitiligo or alopecia of any AE grade | |
| o | Grade 3 infusion-related reaction (first occurrence and in the absence of steroid prophylaxis) that resolves within 6 hours with appropriate clinical management | |
| o | Grade 3 or 4 lymphopenia |
| Page 35 of 92 |
| 6.0 | Dose Delays/Modifications for Adverse Events | |
| 6.1 | Dose Modifications |
It is anticipated that most of the treatment related toxicity on this trial will be caused by carboplatin/etoposide/atezolizumab. Myelosuppression, predominantly neutropenia, will occur frequently; common non-hematologic toxicities include fatigue, nausea, vomiting, and mucositis. In contrast, LB-100 is anticipated to be well tolerated; few toxicities observed in phase I overlapped the known toxicity profile of carboplatin, etoposide and atezolizumab. The following general dose modification rules will, therefore, be used for patients on the LB-100 treatment arm:
If the initiation of a cycle is delayed due to carboplatin/etoposide/atezolizumab toxicity, the LB-100 will also be delayed to begin concurrently with the carboplatin/etoposide/atezolizumab.
If atezolizumab is held then LB-100 should be held as well, as it is a potential immunomodulator
If toxicity is typical of carboplatin/etoposide/atezolizumab and requires dose reductions, the dose of LB-100 should not be reduced.
If the toxicity is attributed specifically to one or two agents (carboplatin, etoposide, atezolizumab), the attributed agents will be dose reduced; otherwise, the doses of all 3 drugs should be reduced.
Patients who require a treatment delay of more than 28 days due to toxicity will be discontinued from the study. An exception is given for tapering of steroids. If a patient must be tapered off steroids used to treat adverse events, atezolizumab may be withheld until steroids are discontinued or reduced to prednisone dose (or dose equivalent) ≤ 10 mg/day.
6.2 Carboplatin/Etoposide Dose Modifications
Two dose reductions of carboplatin and etoposide are allowed. Patients who require dose reductions will not have re-escalation. If grade 3/4 toxicity reoccurs after 2 dose reductions have occurred, the offending agent or agents will be discontinued. If carboplatin, etoposide and atezolizumab must be discontinued due to toxicity, LB-100 will also be discontinued. Patients who require a treatment delay of more than 28 days due to toxicity will be discontinued from the study.
Dose reductions for carboplatin and etoposide are shown in Table 6.2.0
Table 6.2.0 Dose Reductions for Carboplatin & Etoposide
| Dose Level | Carboplatin (AUC) | Etoposide (mg/ m²) | ||
| Starting Dose | 5.0 | 100 x 3 days | ||
| -1 | 4.5 | 75 x 3 days | ||
| -2 | 4.0 | 50 x 3 days |
| Page 36 of 92 |
6.2.1 Hematologic Toxicity
Dose adjustment will be based on the blood count measured on Day 1 (+/- 2 days) of each cycle. No dose modifications will be based on nadir counts. See Table 6.2.1 below.
Table 6.2.1 Dose adjustments for carboplatin and for hematologic toxicity
| Blood Counts | Carboplatin (AUC) | Etoposide (mg/ m²) | ||
|
ANC ≥ 1500/ μL and Platelets ≥100,000/μL |
No dose modification | No dose modification | ||
|
ANC <1500/ μL or Platelets <100,000/ μL |
Delay dosea
Resume with one level dose reduction. Consider the addition of prophylactic G-CSF |
Delay dosea
Resume with one level dose reduction. Consider the addition of prophylactic G-CSF |
||
| Febrile neutropenia (ANC ≤ 1000/ μL and Temp ≥101° F (38.5 °C)] |
Delay doseb
Resume with one level dose reduction. Consider the addition of prophylactic G-CSF |
Delay doseb
Resume with one level dose reduction. Consider the addition of prophylactic G-CSF |
a Check counts at least weekly until ANC ≥1500/ μL and platelets ≥ 100,000/, μL then proceed with Day 1 dose
b Delay dose until the infection is adequately treated and blood counts are ANC ≥1500/ μL and platelets ≥100,000/ μL
6.2.2 Non-Hematologic Toxicity
If grade 3 or 4 non-hematologic toxicity occurs:
| ● | Delay treatment with all drugs | |
| ● | Make an assessment regarding which drug or drugs produced the toxicity | |
| ● | Reevaluate the patient at least once weekly until the toxicity resolves to ≤ grade 1 | |
| ● | Reduce the dose of the offending agent or agents by one dose level | |
| ● | If toxicity is irreversible or has not resolved to ≤ grade 1 after a 3-week treatment delay, the patient should be removed from the study | |
| ● | Creatinine clearance (Cockcroft and Gault formula) should be ≥45 mL/min prior to the start of any cycle. |
6.3 Atezolizumab Dose Holding
There will be no dose reduction for atezolizumab, but patients may temporarily suspend treatment with atezolizumab for up to 4 weeks beyond the last dose if they experience an adverse event that requires a dose to be held. An exception is given for tapering of steroids. If a patient must be tapered off steroids used to treat adverse events, atezolizumab may be withheld until steroids are discontinued or reduced to prednisone dose (or dose equivalent) ≤ 10 mg/day.
| Page 37 of 92 |
6.3.1 Management of Atezolizumab-Specific Adverse Events
Additional tests, such as autoimmune serology or biopsies, should be used to determine a possible immunogenic etiology.
Although most immune-mediated adverse events observed with immunomodulatory agents have been mild and self-limiting, such events should be recognized early and treated promptly to avoid potential major complications. Discontinuation of atezolizumab may not have an immediate therapeutic effect and, in severe cases, immune-mediated toxicities may require acute management with topical corticosteroids, systemic corticosteroids or other immunosuppressive agents.
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
| Abdominal pain | Acute Abdominal pain |
Symptoms of abdominal pain associated with elevations of amylase and lipase, suggestive of pancreatitis, have been associated with administration of other immunomodulatory agents. The differential diagnosis of acute abdominal pain should include pancreatitis.
Appropriate workup should include an evaluation for obstruction, as well as serum amylase and lipase tests. See the guidelines for “Amylase and/or lipase increase” and “Immune- related pancreatitis” elsewhere in this table, as needed.
Right upper-quadrant abdominal pain and/or unexplained nausea or vomiting should be evaluated for potential hepatotoxicity (see the “Hepatotoxicity” guideline elsewhere in this table). |
||
| Adrenal insufficiency | Grade 2+ |
Hold atezolizumab. (symptomatic)
Consider referral of patient to endocrinologist.
Perform appropriate imaging.
Initiate treatment with 1−2 mg/kg/day intravenous methylprednisolone or equivalent and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement. |
||
| Page 38 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
|
If event resolves to Grade 1 or better and patient is stable on replacement therapy (if required) within 4 weeks, taper corticosteroids over ≥1 month and resume atezolizumab.
Permanently discontinue atezolizumab if event does not resolve to Grade 1 or better or patient is not stable on replacement therapy within 4 weeks. |
||||
| Amylase and/or lipase increased | Grade 1 |
Continue atezolizumab.
Monitor amylase and lipase prior to dosing. |
||
| Grade 2 |
Continue atezolizumab.
Monitor amylase and lipase weekly.
For prolonged elevation (e.g., >3 weeks), consider treatment with 10 mg/day oral prednisone or equivalent |
|||
| Grade 3 or 4 |
Hold atezolizumab.
Consider referral of patient to gastrointestinal (GI) specialist.
Monitor amylase and lipase every other day.
If no improvement, consider treatment with 1−2 mg/kg/day oral prednisone or equivalent.
If event resolves to Grade 1 or better within 4 weeks, taper corticosteroids over ≥1 month and resume atezolizumab.
Permanently discontinue atezolizumab if event does not resolve to Grade 1 or better within 4 weeks.
For recurrent events, permanently discontinue atezolizumab. |
|||
| Dermatologic toxicity/rash | Grade 1 | Continue atezolizumab. | ||
| Page 39 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
|
(e.g., maculopapular or purpura) |
Consider topical steroids and/or other symptomatic therapy (e.g., antihistamines). | |||
| Grade 2 |
Continue atezolizumab. Consider dermatologist referral.
Administer topical corticosteroids. |
|||
| Grade 3 |
Hold atezolizumab.
Refer patient to dermatologist. Administer oral prednisone 10 mg or equivalent. If the event does not improve within 48−72 hours, increase dose to 1–2 mg/kg/day or equivalent. Restart atezolizumab if event resolves to Grade 1 or better within 4 weeks.
Permanently discontinue atezolizumab if event does not resolve to Grade 1 or better within 4 weeks. |
|||
| Grade 4 |
Permanently discontinue atezolizumab.
Patient may not resume treatment, regardless of benefit. Otherwise, manage as above. |
|||
|
Persistent and/or severe rash or pruritus, any grade |
A dermatologist should evaluate the event. A biopsy should be performed, unless contraindicated. | |||
| Diarrhea or colitis | Any grade |
Patients should be advised to inform the investigator if any diarrhea occurs, even if it is mild.
All events of diarrhea or colitis should be thoroughly evaluated for other more common etiologies. For events of significant duration or magnitude or associated with signs of systemic inflammation or acute-phase reactants (e.g., increased CRP, platelet count, or bandemia): Perform sigmoidoscopy (or colonoscopy, if appropriate) with colonic biopsy, with three to five specimens for standard paraffin block to check for |
||
| Page 40 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
inflammation and lymphocytic infiltrates to confirm colitis diagnosis. |
||||
| Grade 1 |
Continue atezolizumab.
Initiate symptomatic treatment.
Endoscopy is recommended if symptoms persist for >7 days.
Monitor closely |
|||
| Grade 2 |
Hold atezolizumab.
Initiate symptomatic treatment.
Patient referral to GI specialist is recommended.
For recurrent events or events that persist >5 days, initiate treatment with 1−2 mg/kg/day oral prednisone or equivalent.
If event resolves to Grade 1 or better within 4 weeks, taper corticosteroids over ≥1 month and resume atezolizumab.
Permanently discontinue atezolizumab if event does not resolve to Grade 1 or better within 4 weeks. Resumption of atezolizumab may be considered, after consultation with the trial PI, in patients who are deriving benefit and have fully recovered from the immune-related event. |
|||
| Grade 3 |
Hold atezolizumab.
Refer patient to GI specialist for evaluation and confirmatory biopsy.
Initiate treatment with 1−2 mg/kg/day intravenous methylprednisolone or equivalent and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement. |
|||
| Page 41 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
|
If event resolves to Grade 1 or better within 4 weeks, taper corticosteroids over ≥1 month and resume atezolizumab.
Permanently discontinue atezolizumab if event does not resolve to Grade 1 or better within 4 weeks. Resumption of atezolizumab may be considered, after consultation with the Principal Investigator, in patients who are deriving benefit and have fully recovered from the immune-related event. |
||||
| Grade 4 |
Permanently discontinue atezolizumab. Patient may not resume treatment, regardless of benefit.
Refer patient to GI specialist for evaluation and confirmation biopsy.
Initiate treatment with 1−2 mg/kg/day intravenous methylprednisolone or equivalent and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
If event resolves to Grade 1 or better, taper corticosteroids over ≥1 month. |
|||
| Hepatotoxicity |
Right upper- abdominal pain &/or nausea or vomiting |
Risk of immune-mediated hepatitis. LFTs should be performed immediately, and LFTs should be reviewed before administration of the next dose of study drug. For patients with unexplained elevated LFTs, concurrent medication, viral hepatitis, and toxic or neoplastic etiologies should be considered and addressed, as appropriate.
Symptoms of abdominal pain associated with elevations of amylase and lipase, suggestive of pancreatitis, have been associated with the administration of atezolizumab. The differential diagnosis of acute abdominal pain should also include pancreatitis, as described below. |
||
| Page 42 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
| Grade 1 hepatic event |
Continue atezolizumab.
Monitor LFTs until values resolve to within normal limits. |
|||
| Grade 2 hepatic event, ≤ 5 days |
Continue atezolizumab.
Monitor LFTs more frequently until values resolve to baseline values. |
|||
| Grade 2 hepatic event, > 5 days |
Hold atezolizumab.
Initiate treatment with 1−2 mg/kg/day oral prednisone or equivalent.
If event resolves to Grade 1 or better within 4 weeks, taper corticosteroids over ≥1 month and resume atezolizumab.
Permanently discontinue atezolizumab if event does not resolve to Grade 1 or better within 4 weeks. |
|||
| Grade 3 or 4 hepatic event |
Permanently discontinue atezolizumab.
Consider patient referral to GI specialist for evaluation and liver biopsy to establish etiology of hepatic injury.
Initiate treatment with 1−2 mg/kg/day oral prednisone or equivalent.
If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
If event resolves to Grade 1 or better, taper corticosteroids over > 1 month. Continue atezolizumab. |
|||
| Hyperglycemia | Grade 1 or 2 |
Initiate treatment with insulin if needed.
Monitor for glucose control. |
||
| Grade 3 or 4 | Hold atezolizumab. | |||
| Page 43 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
|
Initiate treatment with insulin.
Monitor for glucose control.
Resume atezolizumab when symptoms resolve and glucose levels are stable. |
||||
| Hyperthyroidism | Grade 1 (asymptomatic) |
TSH > 0.1mU/L and <0.5mU/L: Continue atezolizumab. Monitor TSH every 4 weeks.
TSH < 0.1mU/L: Follow guidelines for symptomatic hyperthyroidism. |
||
| Grade 2+ (symptomatic) |
Hold atezolizumab.
Initiate treatment with anti-thyroid drug such as methimazole or carbimazole as needed.
Consider patient referral to endocrinologist.
Resume atezolizumab when symptoms are controlled and thyroid function is improving.
Permanently discontinue atezolizumab for life-threatening immune-related hyperthyroidism. |
|||
| Hypothyroidism | Grade 1 (asymptomatic) |
Continue atezolizumab.
Start thyroid-replacement hormone. Monitor TSH weekly. |
||
| Grade 2+ | Hold atezolizumab. | |||
| (symptomatic) | ||||
| Start thyroid-replacement hormone. Consider referral to an endocrinologist. | ||||
| Monitor TSH weekly. | ||||
| Page 44 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
| Restart atezolizumab when symptoms are controlled and thyroid function is improving | ||||
| Meningo- | All grades | Permanently discontinue atezolizumab. Patient may not resume treatment, regardless of benefit. | ||
| encepahlitis, | ||||
| immune-related | ||||
| (signs and | Refer patient to neurologist. | |||
| symptoms in | ||||
| absence of an identified alternate etiology) | Initiate treatment with 1−2 mg/kg/day IV methylprednisolone or equivalent and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement. | |||
| If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month. | ||||
| If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent. | ||||
|
Myasthenia gravis and Guillain-Barré syndrome |
All grades |
Permanently discontinue atezolizumab. Patient may not resume treatment, regardless of benefit.
Refer patient to neurologist.
Initiate treatment as per institutional guidelines.
Consider initiation of 1−2 mg/kg/day oral or IV prednisone or equivalent. |
||
| Myocarditis | All grades | Permanently discontinue atezolizumab. Patient may not resume treatment, regardless of benefit. | ||
| Nephritis | Grade 2 |
Withhold atezolizumab.
Refer patient to renal specialist and consider renal biopsy and supportive measures as indicated. Corticosteroids and/or additional immunosuppressive agents should be administered as clinically indicated. |
||
| Page 45 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
| If event resolves to Grade 1 or better within 4 weeks, taper corticosteroids over ≥1 month and resume atezolizumab. | ||||
| Grade 3-4 |
Permanently discontinue atezolizumab.
Refer patient to renal specialist and consider renal biopsy and supportive measures as indicated. Corticosteroids and/or additional immunosuppressive agents should be administered as clinically indicated. |
|||
|
Neuropathy, immune-related
(sensory and/or motor) |
Grade 1 |
Continue atezolizumab.
Evaluate for alternative etiologies. |
||
| Grade 2 |
Hold atezolizumab.
Evaluate for alternative etiologies.
Initiate treatment as per institutional guidelines.
Resume atezolizumab if event resolves to Grade 1 or better within 4 weeks. |
|||
| Grade 3 or 4 |
Permanently discontinue atezolizumab.
Initiate treatment as per institutional guidelines. |
|||
| Ocular event (e.g., uveitis, retinal events | Grade 1 |
Continue atezolizumab.
Patient referral to ophthalmologist is strongly recommended.
Initiate treatment with topical corticosteroid eye drops and topical immunosuppressive therapy.
If symptoms persist, treat as a Grade 2 event. |
||
| Grade 2 | Withhold atezolizumab. | |||
| Page 46 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
|
Patient referral to ophthalmologist is strongly recommended.
Initiate treatment with topical corticosteroid eye drops and topical immunosuppressive therapy.
If event resolves to Grade 1 or better within 4 weeks, taper corticosteroids over ≥1 month and resume atezolizumab. Permanently discontinue atezolizumab if event does not resolve to Grade 1 or better within 4 weeks. |
||||
| Grade 3 or 4 |
Permanently discontinue atezolizumab.
Refer patient to ophthalmologist.
Initiate treatment with 1−2 mg/kg/day oral prednisone or equivalent.
If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month. For Grade 3 AEs, patient may only resume treatment after consultation with the trial PI; for Grade 4, patient cannot resume treatment, regardless of benefit. |
|||
| Pancreatitis, | Grade 2 or 3 | Hold atezolizumab. | ||
| immune related | ||||
| Refer patient to GI specialist. | ||||
| Initiate treatment with 1−2 mg/kg/day intravenous methylprednisolone or equivalent and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement. | ||||
| If event resolves to Grade 1 or better within 4 weeks, taper corticosteroids over ≥ 1 month and resume atezolizumab. | ||||
| Page 47 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
|
Permanently discontinue atezolizumab if event does not resolve to Grade 1 or better within 4 weeks. Patient may only resume treatment after consultation with the trial PI.
For recurrent events, permanently discontinue atezolizumab. Patient may not resume treatment, regardless of benefit. |
||||
| Grade 4 |
Permanently discontinue atezolizumab. Patient may not resume treatment, regardless of benefit.
Refer patient to GI specialist.
Initiate treatment with 1−2 mg/kg/day intravenous methylprednisolone or equivalent and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month. |
|||
| Pulmonary toxicity | All events |
Evaluate thoroughly for other commonly reported etiologies such as pneumonia/infection, lymphangitic carcinomatosis, pulmonary embolism, heart failure, chronic obstructive pulmonary disease (COPD), or pulmonary hypertension. |
||
| Grade 1 |
Continue atezolizumab and monitor closely.
Re-evaluate on serial imaging.
Consider patient referral to a pulmonary specialist.
For recurrent pneumonitis, treat as a Grade 3 or 4 event. |
|||
| Grade 2 | Hold atezolizumab. | |||
| Page 48 of 92 |
| Atezolizumab AE Management and Dose Interruption Guidelines for Specific Toxicities | ||||
| Toxicity | Severity/ Duration | Management | ||
|
Refer patient to pulmonary and infectious disease specialists and consider bronchoscopy or bronchoscopic alveolar lavage (BAL).
Initiate treatment with 1−2 mg/kg/day oral prednisone or equivalent.
If event resolves to Grade 1 or better within 4 weeks, taper corticosteroids over ≥ 1 month and resume atezolizumab. |
||||
| Grade 3 or 4 |
Hold atezolizumab.
Bronchoscopy or BAL is recommended.
Initiate treatment with 1−2 mg/kg/day oral prednisone or equivalent.
If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
If event resolves to Grade 1 or better, taper corticosteroids over ≥1 month. For Grade 3 AEs, patient may only resume treatment after consultation with the Principal Investigator; for Grade 4, patient cannot resume treatment, regardless of benefit. |
|||
6.3.1.1 Systemic Immune Activation
Systemic immune activation is a rare condition characterized by an excessive immune response. Given the mechanism of action of atezolizumab, systemic immune activation is considered a potential risk. Systemic immune activation should be included in the differential diagnosis for patients who, in the absence of an alternative etiology, develop a sepsis-like syndrome after administration of atezolizumab, and the initial evaluation should include the following:
| ● | CBC with peripheral smear | |
| ● | PT, PTT, fibrinogen, and D-dimer | |
| ● | Ferritin | |
| ● | Triglycerides |
| Page 49 of 92 |
| ● | AST, ALT, and total bilirubin | |
| ● | LDH | |
| ● | Complete neurologic and abdominal examination (assess for hepatosplenomegaly) |
6.4 LB-100 Dose Modifications
Two dose reductions of LB-100 are allowed. Re-escalation is allowed once at the discretion of the investigator. Patients with a delay of more than 21 days of LB-100 must be discontinued from study therapy. If grade 3/4 toxicity attributed to LB-100 occurs after 2 previous dose reductions, LB-100 will be discontinued. Patients who are benefiting from treatment may continue carboplatin/etoposide/atezolizumab. Dose reductions of LB-100 are outlined in Table 6.4.0
Table 6.4.0 LB-100 Dose Levels
| Dose Level | LB-100 Dose | |
| -2* | 0.50 mg/m2 | |
| -1 | 0.83 mg/m2 | |
| 1 | 1.25 mg/m2 | |
| 2 | 1.75 | |
| 3 | 2.13 | |
| 4 | 3.10 | |
| *for within patient dose de-escalation only | ||
6.4.1 Hematologic Toxicity
Myelosuppression may infrequently occur with LB-100. Therefore, if grade 3/4 myelosuppression occurs, for the first occurrence the doses of carboplatin and etoposide will be reduced, but LB-100 will stay the same. For the second occurrence of Grade 3/4 myelosuppression LB-100 will be reduced. Atezolizumab will be delayed or discontinued if autoimmune cyctopenias occur. There were no notable adverse events reported in the Phase I trial and we do not expect dose reductions or interruptions.
| Page 50 of 92 |
6.4.2 Non-hematologic Toxicity
The non-hematologic toxicity attributed to LB-100 should be managed as outlined in Table 6.4.2.
Table 6.4.2 Dose adjustments of LB-100
| Toxicity | Management | Dose Reduction | ||
| Injection Site Reaction, grade 3 |
1.) Interrupt LB-100 2.) Administer topical treatment as necessary |
Reduce 1 dose level | ||
| Grade 2 Nephrotoxicity |
1.) Interrupt LB-100 2.) Reexamine patient at least weekly until toxicity improved to ≤ grade 1 |
Prolong infusion time to 2 hours. | ||
| Grade 3 or 4 Nephrotoxicity |
1.) Interrupt LB-100 2.) Reexamine patient at least weekly until toxicity improved to ≤ grade 1 |
Reduce 1 dose level and prolong infusion time to 2 hours. | ||
| Other Grade 2 clinically significant non-hematologic toxicity* |
1.) Interrupt LB-100 2.) Reexamine patient at least weekly until toxicity improved to ≤ grade 1 |
First occurrence: Maintain Dose Second occurrence: Reduce 1 dose level |
||
| Other Grade 3-4 clinically significant non-hematologic toxicity* |
1.) Interrupt LB-100 2.) Reexamine patient at least weekly until toxicity improved to ≤ grade 1 |
Reduce 1 dose level | ||
| Any toxicity requiring a hold of atezolizumab |
1.) Interrupt LB-100 2.) Reexamine patient at least weekly until atezolizumab can be restarted |
Maintain dose level. |
*Alopecia, and clinically insignificant lab abnormalities are examples of things that would not be considered clinically significant
6.5 Pharmacokinetic Studies
6.5.1 Pharmacokinetic Sampling
Plasma for pharmacokinetic (PK) measurements of LB-100, and its major metabolite endothall will be collected in all patients according the sample schedule shown in Table 6.5.1. The sampling schedule allows for determination of LB-100 and endothall PK when LB-100 is given prior to etoposide (Day 1) and when it is given together with etoposide (Day 3). Etoposide PK will also be assessed in patients in the expanded MTD cohort both alone (Day 2) and in combination with LB-100 (Day 3). For measurement of LB-100 and endothall, 5 mL of venous blood will be drawn into a chilled heparin collection tube (sodium or lithium)and kept on ice until the plasma is separated. Plasma will be aliquoted (two aliquots) into appropriately labeled polypropylene tubes (1.8-2 mL cryovials) containing 0.5N NaOH. For every 1.0 mL of plasma aliquoted 0.1 mL of 0.5N NaOH is to be added. Samples will be stored at -70° C until the time of shipment. For measurement of etoposide, an additional 4 mL of venous blood will be drawn into EDTA-containing collection tubes at the times indicated in Table 6.5.1. Tubes will be kept on ice until plasma is separated and aliquoted into appropriately labeled cryovials and stored at < -70°C for subsequent batch analysis.
Procedures for the processing, storage, and shipment of the samples are located in the Laboratory Manual.
| Page 51 of 92 |
(ii) Table 6.5.1: Pharmacokinetic Sample Schedule
|
Study Day |
Draw Time |
One (1) 5 mL heparin tube for LB-100 and endothall |
One (1) 4 mL EDTA tube for etoposide* |
|||
| Day 1 | pre-dose | X | ||||
| immediately at end of LB-100 infusion | X | |||||
| 15 minutes (± 5 minutes) post LB-100 infusion | X | |||||
| 30 minutes (± 5 minutes) post LB-100 infusion | X | |||||
| 1 hour (± 15 minutes) post LB-100 infusion | X | |||||
| 2 hours (± 15 minutes) post LB-100 infusion | X | |||||
| 4 hours (± 30 minutes) post LB-100 infusion and prior to etoposide. | X | |||||
| Day 2 | Pre-treatment (24 hours (± 60 minutes) post LB-100 infusion on day 1) | X | X* | |||
| immediately prior to the end of etoposide infusion | X* | |||||
| 2 hours (± 30 minutes) post etoposide infusion | X* | |||||
| 6 hours (± 30 minutes) post etoposide infusion | X* | |||||
| Day 3 | Pre-treatment [48 hours (± 60 minutes) post LB-100 infusion on day 1 and 24 hours (± 60 minutes) etoposide infusion on day 2] | X | X* | |||
| immediately at end of LB-100 infusion | X | |||||
| 15 minutes (± 5 minutes) post LB-100 infusion | X | |||||
| 30 minutes (± 5 minutes) post LB-100 infusion | X |
| Page 52 of 92 |
| 1 hour (± 15 minutes) post LB-100 and pre etoposide | X | X* | ||||
| 2 hours (± 15 minutes) post LB-100 and immediately prior to end of etoposide infusion | X | X* | ||||
| 3 hours (± 30 minutes) post LB-100 and 1 hours (± 30 minutes) post etoposide | X | |||||
| 4 hours (± 30 minutes) post LB-100 and 2 hours (± 30 minutes) post etoposide | X | X* | ||||
| 8 hours (± 30 minutes) post LB-100 and 6 hours (± 30 minutes) post etoposide | X | X* | ||||
| Day 4 | Post-treatment [24 hours (± 60 minutes) post LB-100 and 22 hours (± 60 minutes) post etoposide on day 3] | X | X* |
*Samples for etoposide PK will be collected only in patients enrolled in the expanded MTD cohort.
6.5.2 Pharmacokinetic Data Analysis
Plasma PK data will be analyzed using both non-compartmental and compartmental methods to derive the relevant secondary PK parameters. Non-compartmental PK methods will be used to determine the parameters (e.g. Cmax, Tmax t1/2, AUC0-t, and CL) for LB-100 and its major metabolite endothall. Compartmental PK analyses of the etoposide data will be performed using ADAPT 5 software (USC Biomedical Simulations Resource, Los Angeles CA), and secondary PK parameters (e.g. CLsys, Vd, t1/2, AUC0-∞) will be determined for each individual. Individual non-compartmental and compartmental PK parameters for each drug and metabolite will be summarized, and potential exposure-response relationships for both safety and efficacy will be assessed.
| 7.0 | Unanticipated Problems and Adverse Event Reporting | |
| 7.1 | Definitions |
| 7.1.1 | Adverse Event |
An adverse event is any untoward medical experience or change of an existing condition that occurs during or after treatment, whether or not it is considered to be related to the protocol intervention.
| Page 53 of 92 |
| 7.1.2 | Serious Adverse Event (SAE) |
A serious adverse event is any expected or unexpected adverse events that result in any of the following outcomes:
| ● | Death | |
| ● | Is life-threatening experience (places the subject at immediate risk of death from the event as it occurred) | |
| ● | Unplanned hospitalization (equal to or greater than 24 hours) or prolongation of existing hospitalization | |
| ● | A persistent or significant disability/incapacity | |
| ● | A congenital anomaly/birth defect | |
| ● | Secondary malignancy* | |
| ● | Any other adverse event that, based upon appropriate medical judgment, may jeopardize the subject’s health and may require medical or surgical intervention to prevent one of the outcomes listed above (examples of such events include allergic bronchospasm requiring intensive treatment in the emergency room or at home, blood dyscrasias of convulsions that do not result in inpatient hospitalization, or the development of drug dependency or drug abuse). |
*Modified from 21 CFR 312.32
| 7.1.3 | Unanticipated Problems Involving Risks to Subjects or Others |
An unanticipated problem is any incident, experience, or outcome that meets all three of the following criteria:
| 1. | Unexpected (in terms of nature, severity, or frequency) given the following: a) the research procedures described in the protocol-related documents such as the IRB approved research protocol, informed consent document or Investigator Brochure (IB); and b) the characteristics of the subject population being studied; AND | |
| 2. | Related or possibly related to participation in the research (possibly related means there is a reasonable possibility that the incident, experience, or outcomes may have been caused by the drugs, devices or procedures involved in the research); AND | |
| 3. | Suggests that the research places subjects or others at greater risk of harm (including physical, psychological, economic, or social harm) than previously known or recognized. |
| 7.1.4 | Adverse Events of Special Interest (AESI) |
Specific adverse events, or groups of adverse events, will be followed as part of standard safety monitoring activities. These events, regardless of seriousness, will be reported.
| Page 54 of 92 |
| 7.1.4.1 | Study specific AESIs |
Currently, there are no specific adverse events for this study.
| 7.1.5 | Pregnancy |
Any pregnancy diagnosed during the study, or that occurs within 30 days after stopping study medication, must be reported immediately to the Investigator. Pregnancy, in and of itself, is not regarded as an adverse event, unless there is a suspicion that study medication may have interfered with the effectiveness of a contraceptive medication. If the patient becomes pregnant while on-study, the study drug should be immediately discontinued. Pregnancy information about a female patient or female partner of a male patient should be reported immediately from the time the Investigator first becomes aware of a pregnancy or its outcome.
Any pregnancy complication, spontaneous abortion, elective termination of a pregnancy for medical reasons, the outcome of stillbirth, congenital anomaly/birth defect, or serious adverse event in the mother will be recorded as an SAE and will be reported as described in Section 7.3.
7.2 Assessment of Adverse Events
The site Investigator will be responsible for determining the event name, assessing the severity (i.e. grade), expectedness, and attribution of all adverse events.
| 7.2.1 | Assessment of Adverse Event Name and Grade |
Adverse events will be characterized using the descriptions and grading scales found in the most recent version of CTCAE version 5.0. A copy of the scale can be found at https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm. The determination of severity for all other events not listed in the CTCAE version 5.0. should be made by the investigator based on medical judgment and the severity categories of Grade 1 to 5 as defined below:
| ● | Grade 1 (mild) – An event that is usually transient and may require only minimal treatment or therapeutic intervention. The event does not generally interfere with usual activities of daily living. | |
| ● | Grade 2 (moderate) – An event that is usually alleviated with additional specific therapeutic intervention. The event interferes with usual activities of daily living, causing discomfort but poses no significant or permanent risk of harm to the subject. | |
| ● | Grade 3 (severe) – An event that requires intensive therapeutic intervention. The event interrupts usual activities of daily living, or significantly affects the clinical status of the subject. | |
| ● | Grade 4 (life threatening) – An event, and/or its immediate sequelae, that is associated with an imminent risk of death or with physical or mental disabilities that affect or limit the ability of the subject to perform activities of daily living (eating, ambulation, toileting, etc.). | |
| ● | Grade 5 (fatal) – Death (loss of life) as a result of an event. |
| Page 55 of 92 |
| 7.2.2 | Assessment of Attribution |
The following definitions will be used to determine the causality (attribution) of the event to the study agent or study procedure.
| ● | Unrelated – The event is clearly related to other factors such as the participant’s clinical state, other therapeutic interventions, or concomitant medications administered to the participant. | |
| ● | Unlikely – The event is doubtfully related to the investigational agent(s). The event was most likely related to other factors such as the participant’s clinical state, other therapeutic interventions, or concomitant drugs. | |
| ● | Possible – The event follows a reasonable temporal sequence from the time of drug administration, but could have been produced by other factors such as the participant’s clinical state, other therapeutic interventions, or concomitant drugs. | |
| ● | Probable – The event follows a reasonable temporal sequence from the time of drug administration, and follows a known response pattern to the study drug. The event cannot be reasonably explained by other factors such as the participant’s clinical state, therapeutic interventions, or concomitant drugs. | |
| ● | Definite – The event follows a reasonable temporal sequence from the time of drug administration, follows a known response pattern to the study drug, cannot be reasonably explained by other factors such as the participant’s condition, therapeutic interventions, or concomitant drugs, AND occurs immediately following study drug administration, improves upon stopping the drug, or reappears on re-exposure. |
| 7.2.3 | Assessment of Expectedness |
The following definitions will be used to determine the expectedness of the event:
| ● | Unexpected – An adverse event is unexpected if it is not listed in the investigator’s brochure and/or package insert; is not listed at the specificity or severity that has been observed; is not consistent with the risk information described in the protocol and/or consent; is not an expected natural progression of any underlying disease, disorder, condition, or predisposed risk factor of the research participant experiencing the adverse event. *Modified from 21 CFR 312.32 (a) | |
| ● | Expected – An adverse event is expected if it does not meet the criteria for an unexpected event, OR is an expected natural progression of any underlying disease, disorder, condition, or predisposed risk factor of the research participant experiencing the adverse event. |
| 7.3 | Reporting of Adverse Events |
| 7.3.1 | Routine Reporting of Non-Serious Adverse Events |
Routine AE recording will occur via data entry into the study eCRF. Recording of adverse events will begin once the patient is consented and will continue until 30 days after last study drug. Adverse events will be monitored by the Protocol Management Team (PMT). Adverse events that do not meet the criteria of serious OR are not unanticipated problems do not require expedited reporting. AEs reported through expedited processes (i.e. reported to the IRB, DSMC, FDA, etc.) must also be reported in routine study data submissions.
| Page 56 of 92 |
| 7.3.2 | Expediting Reporting Requirements of SAEs and UPs |
Adverse events that meet the criteria of serious OR are unanticipated problems will be reported according to the approved City of Hope’s Institutional policy via the AE/UP reporting form in iRIS. Reportable serious adverse events must be followed until the event is resolved, stabilized, or determined to be irreversible by the investigator. Follow-up SAE reports must be submitted for all events that require expedited reporting when the status of the event changes and until the resolution or stabilization of the event.
| 7.3.3 | Additional AE Reporting Requirements |
| 7.3.3.1 | Reporting to the FDA |
The study PI (or designee) will be responsible for contacting the Office of IND Development and Regulatory Affairs (OIDRA) at COH to ensure prompt reporting of safety reports to the FDA. OIDRA will assist the PI with the preparation of the report and submit the report to the FDA in accordance with the approved City of Hope’s Institutional policy. (Appendix D).
Serious Adverse Events meeting the requirements for expedited reporting to the Food and Drug Administration (FDA), as defined in 21 CFR 312.32, will be reported as an IND safety report using the MedWatch Form FDA 3500A for Mandatory Reporting.
The criteria that require reporting using the MedWatch 3500A are:
| ● | Any unexpected fatal or life threatening adverse experience associated with use of the drug must be reported to the FDA no later than 7 calendar days after initial receipt of the information [21 CFR 312.32 (c) (2)] | |
| ● | Any adverse experience associated with use of the drug that is both serious and unexpected must be submitted no later than 15 calendar days after initial receipt of the information [21 CFR 312.32 (c) (1)] | |
| ● | Any follow-up information to a study report shall be reported as soon as the relevant information becomes available. [21 CFR 312.32 (d) (3)] |
In addition, the study PI will submit annually within 60 days (via COH OIDRA) of the anniversary date of when the IND went into effect, an annual report to the FDA which is to include a narrative summary and analysis of the information of all FDA reports within the reporting interval, a summary report of adverse drug experiences, and history of actions taken since the last report because of adverse drug experiences.
| 7.3.3.2 | Reporting to Lixte Biotechnology |
All serious adverse events, AESIs (initial and follow-up information) and pregnancies will be reported by the study PI to Lixte Biotechnology (via Theradex Oncology) within 24 hours of becoming aware of the event.
| Theradex Safety Desk | |
| Telephone: (609) 799-7580 | |
| Fax: (609) 799-1567 | |
| Email: SafetyDeskUS@Theradex.com |
| Page 57 of 92 |
| V. | 8.0 Agent Information and Risks |
(i) 8.1 LB-100
8.1.1 Description
LB-100 has been shown in a Phase I clinical trial of solid tumors that it can be well tolerated at doses associated with partial response and stable disease in patients who had failed multiple treatment regimens (Chung 2016). This and other preclinical studies have shown that LB-100 has efficacy alone and in combination with standard cytotoxic chemotherapy and/or radiation without enhancing toxicity.
Chemistry: LB-100 (3-[(4-Methylpiperazin-1-yl)carbonyl]-7-oxabicyclo[2.2.1]heptane-2-carboxylic acid], NSC D753810 is a water-soluble small molecule inhibitor of PP2A.
Mechanism of Action: LB-100 inhibits PP2A which leads to hyperactivation of Ras signaling and the stabilization of c-Myc during the G2 phase that eventually drives cells into mitosis through the accumulation of cyclin E: Cdk1 complexes.
Human Toxicology: As of April 2020, 29 people have received LB-100. The following side effects have been seen: >25% of patients: Fatigue. > 10% but < 25% of patients: Aspartate aminotransferase increased, Blood creatinine increased, Headache, Hypernatraemia, Hypoalbuminaemia, Nausea, Proteinuria, Pyrexia. <10% of patients: Abdominal discomfort, Abdominal distension, Accelerated hypertension, Alanine aminotransferase increased, Anaemia, Arthralgia, Blood alkaline phosphatase increased, Blood urea increased, Candidiasis, Chest pain, Chills, Constipation, Creatinine renal clearance, Decreased appetite, Dermatitis acneiform, Diarrhoea, Dizziness, Dyspnoea, Ejection fraction decreased, Electrocardiogram qt prolonged, Gait disturbance, Gastrointestinal disorder, Generalised oedema, Gingival pain, Hypercalcaemia, Hyperkalaemia, Hypertension, Hypoaesthesia, Hypokinesia, Hyponatraemia, Hypotension, Hypoxia, Insomnia, Lymphocyte count decreased, Mucosal inflammation, Muscle twitching, Muscular weakness, Neuropathy peripheral, Neutropenia, Oedema, Oedema peripheral, Pain of skin, Peripheral coldness, Peripheral sensory neuropathy, Platelet count decreased, Pleural effusion, Sinus tachycardia, Tachypnoea, Tremor, Vomiting, Weight decreased.
| 8.1.2 | Pharmacology – Handling, Storage, Dispensing and Disposal |
LB-100 will be supplied as a sterile solution for intravenous administration. LB-100 is to be stored at -20C (allowable range -25C to -10C). Each vial contains 10 mL of LB-100 at a concentration of 1 mg/ml. The proper dose is drawn up in a sterile syringe and added to 50 mL of normal saline (0.9%). Following dilution in normal saline, LB-100 should be administered within 4 hours. On Days 1 and 3 of each cycle, LB-100 will be infused over 15 minutes ± 5 minutes.
8.2 Carboplatin
| 8.2.1 | Description |
Carboplatin is an analog of cisplatin that has a more favorable toxicity profile (Ruckdeschel 1994). It interacts with DNA and forms both intra- and interstrand links. The most commonly observed side effects include thrombocytopenia, neutropenia, leukopenia, and anemia. Like other platinum-containing compounds, carboplatin may induce anaphylactic-type reactions such as facial edema, wheezing, tachycardia, and hypotension that may occur within a few minutes of drug administration. These reactions may be controlled with adrenaline, corticosteroids, or antihistamines.
| Page 58 of 92 |
Chemistry: Carboplatin (carboplatin for injection or platinum diamine [1,1-cyclobutane-decarbozxylate (2—0,0’)-,(SP-4-2)] is a platinum compound used as a chemotherapeutic agent.
Mechanism of Action: Carboplatin undergoes activation inside cells and forms reactive platinum complexes that cause the intra- and inter-strand cross-linkage of DNA molecules within the cell. This modifies the DNA structure and inhibits DNA synthesis. This may affect a cell in all the phases of its cycle.
| 8.2.2 | Toxicology |
Side effects of carboplatin (CBDCA) include myelosuppression, nausea, vomiting, abdominal pain, diarrhea, and constipation. Other toxicities include allergic reaction (including hypersensitivity, i.e., rash, urticaria, erythema, pruritus, bronchospasm and hypotension), peripheral neuropathy, paresthesia, loss of hair, hearing loss, visual disturbances, and change in taste. Serum creatinine elevations and blood urea elevations have occurred as well as abnormal liver function tests and decreased serum electrolyte values. Although rare, pain, asthenia, cardiovascular, respiratory, genitourinary, and mucosal side effects have occurred in some patients. Cancer-associated hemolytic uremic syndrome has been reported rarely. The renal effects of nephrotoxic compounds may be potentiated by carboplatin. Carboplatin is contraindicated in patients with a history of severe allergic reactions to cisplatin or other platinum containing compounds or mannitol. This drug should not be used in patients with severe bone marrow depression or significant bleeding. The occurrence of acute leukemia has been reported rarely in patients treated with anthracycline/alkylator combination chemotherapy.
Pregnancy and Lactation: Carboplatin may cause fetal harm; therefore, women of childbearing potential should be advised to avoid becoming pregnant.
| 8.2.3 | Pharmacology – Handling, Storage, Dispensing and Disposal |
Carboplatin is commercially available and will not be provided by the study sponsor. Carboplatin should be stored at room temperature (15C to 25C) and protected from light. Once diluted, it should be stored at room temperature or refrigerated. Since no antibacterial preservatives are contained in the formulation, it is recommended that any carboplatin solution be discarded after 8 hours from the time of dilution if stored at room temperature or after 24 hours if stored under refrigeration.
8.3 VP-16 (Etoposide)
| 8.3.1 | Description |
Etoposide is a semisynthetic derivative of podophyllotoxin that exhibits cytostatic activity in vitro by preventing cells from entering mitosis or by destroying them at a premitotic stage. Etoposide interferes with the synthesis of DNA and appears to arrest human lymphoblastic cells in the late S-G2 phases of the cell cycle. The most commonly observed side effects include leukopenia and thrombocytopenia. Etoposide is indicated in combination with other antineoplastics in the treatment of SCLC, NSCLC, malignant lymphoma, and testicular malignancies (approved indications may vary depending on the specific country). Etoposide is also used in clinical studies against many other types of cancer including head and neck, brain, bladder, cervical, and ovarian.
Chemistry: VP-16 is a semi-synthetic podophyllotoxin derivative from the plant podophyllum pletatum, and has antineoplastic properties in experimental animals and in man. The empiric formula C29H32O13 has a molecular weight of 588.
Mechanism of Action: The epipodophyllotoxins exert phase specific spindle poison activity with metaphase arrest, but in contrast to the vinca-alkaloids, have an additional activity of inhibiting cells from entering mitosis. Suppression of tritiated thymidine, uridine, and leucine incorporation in human cells in tissue culture suggests effects against DNA, RNA, and protein synthesis.
| Page 59 of 92 |
| 8.3.2 | Toxicology |
Animal Toxicology: The predominant toxicities of VP-16 in animal studies involve the hematopoietic system, with toxicity to the liver and GI tract occurring only at doses producing profound myelosuppression. Anemia, leukopenia and lymphoid involution occur in mice, rats and monkeys. Acute toxicity investigations have been complicated by the toxicity of the solvent system. The LD-50 of the solvent plus drug approached that of the solvent alone.
Immunosuppressive effects occur within an inhibition of antibody production in mice and monkeys, and prevention of experimental allergic encephalomyelitis in rats (cell-mediated immunity).
Human Toxicology: Reversible myelotoxicity has been uniformly observed to be the major toxicity of VP- 16 and to represent the only clinically significant side effect. Following a single IV injection, peak myelotoxicity occurs at seven to nine days. Following daily IV injections for five to seven days, myelotoxicity is maximal between 12-16 days from the initiation of therapy. Bone marrow suppression is mainly manifested as granulocytopenia, with thrombocytopenia and anemia occurring to a lesser extent. Gastrointestinal toxicities including transient modest nausea, vomiting, and diarrhea are common. Other reactions could include aftertaste, rash, pigmentation, pruritus, abdominal pain, constipation, and dysphagia. Occasional alopecia is reported. VP-16 does not produce phlebitis, or nephrotoxicity. Rarely, anaphylactic-like reactions have been reported, as well as, hypotension. Hypotension can be managed by infusing the drug over at least a 30-minute period. Occasionally, chills, fever, peripheral neurotoxicity, stomatitis, hepatotoxicity, transient cortical blindness and radiation recall dermatitis may be a result of VP- 16 administration. The occurrence of acute leukemia has been reported rarely in patients treated with VP- 16 in association with other antineoplastic agents. VP-16 can cause fetal harm when administered to pregnant women.
Pregnancy and Lactation: Etoposide can cause fetal harm when administered to a pregnant woman. Etoposide has been shown to be teratogenic in mice and rats. In these studies, etoposide caused dose-related material toxicity, embryotoxicity and teratogenicity. Fetal abnormalities included decrease weight, major skeletal abnormalities, exencephaly, encephalocele, anophthalmia, and retarded ossification. No information is available on excretion of this drug in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants, it is recommended that nursing be discontinued.
| 8.3.3 | Pharmacology – Handling, Storage, Dispensing and Disposal. |
Etoposide is commercially available and will not be provided by the study sponsor. Etoposide should be stored at room temperature (15C to 25C) and protected from light. It should not be administered by intravenous push. Sites must not store any unused portion of a vial for future use and must discard unused portions of product. Follow reconstitution and dilution instructions in the package insert.
| Page 60 of 92 |
8.4 Atezolizumab
| 8.4.1 | Description |
Atezolizumab is an Fc-engineered, humanized, non-glycosylated IgG1 kappa monoclonal antibody that binds to and blocks programmed death-ligand 1 (PD-L1) approved for certain types of urothelial cancer and non-small cell lung cancer (NSCLC). It is indicated for the treatment of locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy, or who are ineligible for cisplatin and have high tumor expression of PD-L1, or who are ineligible for other platinum-containing chemotherapy regardless of tumor PD-L1 expression; it is also indicated for the treatment of metastatic NSCLC. In an ongoing multicenter, randomized trial in previously untreated patients with metastatic urothelial carcinoma who are eligible for platinum-containing chemotherapy (IMvigor130), patients with PD-L1 expression of less than 5% had decreased survival with atezolizumab monotherapy compared to those who received platinum-based chemotherapy; the monotherapy arm of this trial was closed to accrual for patients with low PD-L1 expression upon the recommendation of the independent Data Monitoring Committee. Treatment with atezolizumab may result in severe immune-related adverse reactions requiring interruption or discontinuation of therapy, as well as treatment with high-dose corticosteroids. Infusion-related reactions may also occur.
Chemistry: Atezolizumab is an Fc-engineered, humanized, non-glycosylated IgG1 kappa monoclonal antibody that inhibits programmed death ligand 1 (PD-L1) interactions with the PD-1 and B7.1 receptors.
Mechanism of Action: Atezolizumab is an Fc-engineered, humanized, non-glycosylated IgG1 kappa monoclonal antibody that inhibits programmed death ligand 1 (PD-L1) interactions with the PD-1 and B7.1 receptors. PD-L1 may be expressed on tumor cells and/or tumor-infiltrating immune cells, and can contribute to inhibition of the anti-tumor immune response in the tumor microenvironment; PD-1 and B7.1 receptors are found on T-cells and antigen-presenting cells. The PD-1 pathway regulates the balance between T-cell activation and protection of healthy tissues from immune-mediated damage. In cancer, the PD-1 pathway is thought to play an important role in the interaction of tumor cells with the host immune response. Binding of PD-L1 to the PD-1 and B7.1 receptors suppresses cytotoxic T-cell activity, T-cell proliferation, and cytokine production; PD-L1 expression in a tumor cell may provide adaptive immune resistance and lead to poor outcomes. Atezolizumab binds to PD-L1 and prevents its interaction with both PD-1 and B7.1 receptors, releasing the PD-L1/PD-1 mediated inhibition of an anti-tumor immune response without inducing antibody-dependent cellular cytotoxicity. In syngeneic mouse tumor models, blocking PD-L1 activity resulted in decreased tumor growth.
| 8.4.2 | Toxicology |
Animal Toxicology: In animal models, inhibition of PD-L1/PD-1 signaling increased the severity of some infections 404 and enhanced inflammatory responses. M. tuberculosis-infected PD-1 knockout mice exhibit 405 markedly decreased survival compared with wild-type controls, which correlated with increased 406 bacterial proliferation and inflammatory responses in these animals. PD-L1 and PD-1 knockout 407 mice and mice receiving PD-L1 blocking antibody have also shown decreased survival following 408 infection with lymphocytic choriomeningitis virus.
Human Toxicology:
Urothelial Carcinoma: Atezolizumab was investigated in Study 1, a multicenter, open-label, two-cohort trial that included patients with locally advanced or metastatic urothelial carcinoma. In Cohort 2 of Study 1, 310 patients with locally advanced or metastatic urothelial carcinoma who had disease progression during or following a platinum-containing chemotherapy regimen or who had disease progression within 12 months of treatment with a platinum-containing neoadjuvant or adjuvant chemotherapy regimen were treated with atezolizumab. This study excluded patients who had: a history of autoimmune disease, active or corticosteroid-dependent brain metastases, administration of a live, attenuated vaccine within 28 days prior to enrollment, or administration of systemic immunostimulatory agents or systemic immunosuppressive medications. Patients received an intravenous infusion of 1200 mg of atezolizumab every 3 weeks until unacceptable toxicity or either radiographic or clinical progression. Tumor response assessments were conducted every 9 weeks for the first 54 weeks and every 12 weeks thereafter. Major efficacy outcome measures included confirmed objective response rate (ORR) as assessed by independent review facility (IRF) using Response Evaluation Criteria in Solid Tumors (RECIST v1.1) and duration of response (DoR).
| Page 61 of 92 |
In this cohort, the median age was 66 years, 78% were male, 91% patients were Caucasian. Twenty-six percent had non-bladder urothelial carcinoma and 78% of patients had visceral metastases. Sixty-two percent of patients had an ECOG score of 1 and 35% of patients had a baseline creatinine clearance of <60 mL/min. Nineteen percent of patients had disease progression following prior platinum-containing neoadjuvant or adjuvant chemotherapy. Forty-one percent of patients had received ≥ 2 prior systemic regimens in the metastatic setting. Seventy-three percent of patients received prior cisplatin, 26% had prior carboplatin, and 1% were treated with other platinum-based regimens.
Tumor specimens were evaluated prospectively using the Ventana PD-L1 (SP142) Assay at a central laboratory, and the results were used to define subgroups for pre-specified analysis. Of the 310 patients, 32% were classified as having PD-L1 expression of ≥ 5% (defined as PD-L1 stained tumor-infiltrating immune cells [ICs] covering ≥ 5% of the tumor area). The remaining, 68% of patients, were classified as having PD-L1 expression of <5% (PD-L1 stained tumor-infiltrating ICs covering <5% of the tumor area).
Confirmed ORR in all patients and the two PD-L1 subgroups are summarized in Table 8.4.2 below. The median follow-up time for this cohort was 14.4 months. In 59 patients with disease progression following neoadjuvant or adjuvant therapy, the ORR was 22.0% (95% CI: 12.3, 34.7%).
Table 8.4.2 Overall Response by PD-L1 Expression
| All Patients | PD-L1 Expression Subgroups | ||||
| N=310 |
PD-L1 expression of <5% in ICs1 (N=210) |
PD-L1 expression of ≥5% in ICs1 (N=100) |
|||
| Number of IRF- assessed Confirmed Responders | 46 | 20 | 26 | ||
| ORR % (95% CI) | 14.8% (11.1, 19.3) | 9.5% (5.9, 14.3) | 26.0% (17.7, 35.7) | ||
| Complete Response (CR) (%) | 5.5% | 2.4% | 12.0% | ||
| Partial Response (PR) (%) | 9.4% | 7.1% | 14.0% | ||
| Median DoR, months (range) | NR (2.1+, 13.8+) | 12.7 (2.1+, 12.7) | NR (4.2, 13.8+) | ||
|
NR = Not reached + Denotes a censored value 1 PD-L1 expression in tumor-infiltrating immune cells (ICs) |
|||||
| Page 62 of 92 |
| 8.4.3 | Pharmacology – Handling, Storage, Dispensing and Disposal |
Atezolizumab is commercially available and will not be provided by the study sponsor. Atezolizumab is a sterile, preservative-free, and colorless to slightly yellow solution for intravenous injection supplied as a carton containing one 1200 mg/20 mL single-dose vial (NDC 50242-917-01). Store vials under refrigeration at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not freeze. Do not shake.
9.0 Correlative/Special Studies
The main objectives of the monitoring will be to characterize changes in T cell subsets, monocytes, and other immune cell subsets that can be correlated with the clinical outcome. We will analyze by both single cell RNA sequencing (scRNAseq), with validation of immune cell classification on a subset of samples using multicolor flow cytometry. These studies will measure the following:
| 1. | The relative abundance of immune cell populations before, during and after treatment. | |
| 2. | The signaling states that evolve in immune cells during treatment and that differentiate responders from non-responders. |
We expect to see differences in abundance effector memory T cell populations and classical monocytes before and during treatment. We also expect the pathway analysis using the scRNAseq data to identify those signaling changes that occur during treatment and that distinguish responsive patients. Samples collected in this clinical trial will allow to determine more precisely at what time point the immunological effects are the most significant.
| 1. | Peripheral blood collected at baseline (screening) and pre-treatment of Cycles 1, 4, 7, 10 and EOT. | |
| 2. | Samples will be collected in 10 ml BD Vacutainer CPT tubes. | |
| 3. | Sample Processing: |
CPT tubes will be processed using the following method: Following collection, samples can be stored in an upright position for up two hours at room temperature. Remix the blood samples immediately following collection and prior to centrifugation by gently inverting the tubes 8-10 times. Centrifuge samples at room temperature in a horizontal rotor (swing-out head) at 820g for 10 min at room temperature, with the centrifuge brake off.
For plasma: Transfer 1mL aliquots of the plasma to sterile 2mL microtubes. Take care to avoid any buffy coat layer in this step. Label each aliquot by the order in which it was isolated, 1 thru n for n aliquots. Centrifuge plasma aliquots in a bench top centrifuge at 16,000-20,000g (maximum speed) for 10 min to pellet any remaining cellular debris. Carefully transfer 1ml aliquots of supernatant from plasma to sterile 2 mL screw-capped microtubes. Freeze plasma aliquots at -80 °C or LN2 freezer.
For PBMC/buffy coat: Centrifuge 15 min at 300 RCF. Aspirate as much supernatant as possible without disturbing the cell pellet. Resuspend cell pellet by gently tapping the tube with index finger. Add 10 ml of PBS. Cap tubes and mix cells by inverting tube 5 times. Centrifuge 10 minutes at 300 RCF. Aspirate as much supernatant as possible without disturbing cell pellet. Resuspend cell pellet by gently tapping the tube with index finger. Add 2 ml of Recovery Cell Culture freezing Medium (Life Technology) or 10% DMSO, 40% FBS and 50% RPMI. Transfer cells with a Pasteur pipette to a 1.5ml cryotube. Add 2-propanol at RT into Nalgene Cryofreezing Container. Place the cryovials into the container. Put the freezing container into -80°C freezer for a minimum of 12 hours.
During the freezing time avoid opening the freezer in order to avoid shaking the cryovials or raising the freezer’s temperature. If Nalgene Cryofreezing containers are not available, place the cryovials in a Styrofoam tube container (they are supplied with the 15ml conical tubes) to reduce direct contact with cold surfaces and to slow the rate of freezing. Put the freezing container into -80°C freezer for a minimum of 12 hours. During the freezing time avoid opening the freezer in order to avoid shaking the cryovials or raising the freezer’s temperature.
All samples will be sent to the Viable Cell Biorepository.
| Page 63 of 92 |
| VI. |
10.0 Study
Calendar |
|
Induction=4 cycles |
w/in # days of start |
One Cycle=21 Days | |||||
|
Pre-Study |
Days 1& 3,Week 1 |
Day 1, Week 2 |
Day 1, Week 3 |
Off Study | Follow- up | ||
| LB-100, Atezolizumab, Carboplatin, Etoposide (Induction Cycles 1-4) | X see section 5.1 | ||||||
| LB-100, Atezolizumab, (Maintenance Cycle 5 and beyond) | X see section 5.1 | ||||||
| Informed consent | X | ||||||
| Demographics | X | ||||||
| Medical history | X | ||||||
| Concurrent meds | 14 | X | |||||
| Physical exam | 14 | X | Day 1 only | X | X6 | ||
| Vital sign, Weight | 14 | X | Day 1 only | X | X6 | ||
| Height | X | ||||||
| Performance Status | 14 | X | Day 1 only | X | X6 | ||
| CBC w/diff, plts | 14 | X | Day 1 only | X | X | X | X6 |
| Serum chemistry | 14 | X | Day 1 only | X | X | X | X6 |
| Urinalysis | 14 | X | Day 1 only | X | X | ||
| TSH, free T3, free T4 | 14 | X | Day 1 only | ||||
| EKG (triplicate) 8 | 14 | X | Cycles 1&2 only7 | X8 | X8 | X | |
| Adverse event evaluation | ——————X—————— | ||||||
| Tumor measurements (RECIST)1,2 | 28 | X | Measurements repeated every 6-8 weeks (± 7 days)1 | X5 | |||
| Radiologic evaluation | 28 | X | Should be performed every 6-8 weeks (± 7 days) | X5 | |||
| B-HCG | 14 | X | |||||
| PK | Cycle3 1 | ||||||
| Blood for correlatives | X4 |
Cycles4 1, 4, 7 & 10 |
X4 | ||||
| Survival Follow-up | X | ||||||
| 1 | Documentation (radiologic) must be provided for patients removed from study for progressive disease. | |
| 2 | Appendix B | |
| 3 | See Section 6.5.1. Cycle 1 only. Collect blood in a 5 mL chilled heparin tube Day 1: pre-infusion, end of infusion, and 15 minutes, 30 minutes, 1, hour, 2 hours, 4 hours and 8 hours post. Day 2: pre-infusion. |
| Page 64 of 92 |
| 4 | See Section 9.1 Collect peripheral blood in a 10 mL BD Vacutainer CPT tube at screening, and pre-treatment on Day 1 of Cycles 1, 4,7, 10 and off treatment. | |
| 5 | For patients that come off study for reasons other than progressive disease. 6 30 Days post last study drug | |
| 7 | End of LB-100 infusion | |
| 8 | As clinically indicated throughout the study |
11.0 Endpoint Definitions
11.1 Primary endpoints:
| ● | Determine recommended phase II dose (RP2D) of the combination using DLT (Definition Section 5.7) during the first cycle as assessed by CTCAE version 5.0. |
11.2 Secondary endpoints:
| ● | Objective response rate (ORR) as defined by RECIST v1.1 (Appendix B) | |
| Patients who respond to treatment and die without PD (including death from study disease), duration of response will be censored at the date of the last objective progression-free disease assessment. For responding patients not known to have died as of the data cut-off date and who do not have PD, duration of response will be censored at the last progression-free assessment date. | ||
| For responding patients who receive subsequent anticancer therapy (after discontinuation from all study treatment excluding PCI) prior to disease progression, duration of response will be censored at the date of last progression-free assessment prior to the initiation of post discontinuation anticancer therapy | ||
| ● | Duration of overall response as defined by RECIST v1.1 (Appendix B) | |
| ● | Safety and Adverse events by assessed by CTCAE version 5.0 | |
| ● | Progression-free survival (PFS) as defined by RECIST v1.1 (Appendix B) | |
| ● | Overall survival, which is defined as the time from the date of study enrollment to the date of death from any cause. For patients who are still alive as of the data cutoff date, OS time will be censored on the date of the patient’s last contact (last contact for patients in post discontinuation is last known alive date in mortality status). |
11.3 Exploratory endpoints
| ● | The pharmacokinetics (PK) of LB100 and its metabolite endothall. | |
| ● | The relative abundance of immune cell populations before, during and after treatment. | |
| The signaling states that evolve in immune cells during treatment and that differentiate responders from non-responders. |
12.0 Data Handling, Data Management, Record Keeping
12.1 Source Documents
Source documents are original documents, data, and records (e.g., medical records, pharmacy dispensing records, recorded data from automated instruments, laboratory data) that are relevant to the clinical trial. The investigator or their designee will prepare and maintain adequate and accurate source documents. These documents are designed to record all observations and other pertinent data for each patient enrolled in this clinical trial. Source documents must be adequate to reconstruct all data transcribed onto the case report forms.
| Page 65 of 92 |
12.2 Data Capture Methods and Management
Data for this trial will be collected using City of Hope’s electronic capture system that is compliant with 21 CFR Part 11. Study personnel will enter data from source documents corresponding to a subject’s visit into the protocol-specific electronic Case Report Form (eCRF).
12.3 Case Report Forms/Data Submission Schedule
Study personnel will enter data from source documents corresponding to a subject’s visit into the protocol- specific electronic Case Report Form (eCRF) when the information corresponding to that visit is available.
The investigator is responsible for all information collected on subjects enrolled in this study. All data collected during this study must be reviewed and verified for completeness and accuracy by the investigator. All case report forms must be completed by designated study personnel. The completed case report forms must be reviewed, signed and dated by the Investigator or designee in a timely fashion.
All data will be collected using electronic data collection, stored as indicated in Section 12.2, and will be submitted according to the timelines indicated in Table 12.3.1.
Table 12.3.1: Data Submission Schedule
| Form | Submission Timeline | |
| Eligibility Checklist | Complete prior to registration | |
| On Study Forms | Within 14 calendar days of registration. | |
| Baseline Assessment Forms | Within 14 calendar days of registration. | |
| Treatment Forms | Within 10 calendar days of treatment administration. | |
| Adverse Event Report Forms |
Window period: Within 7 calendar days of AE assessment/notification. Post-window period: Within 10 calendar days of AE assessment/notification. |
|
| Response Assessment Forms | Within 10 calendar days of the response assessment. | |
| Other Assessment Forms (concomitant medications) | Within 10 calendar days of the assessment. | |
| Off Treatment/Off Study Forms | Within 10 calendar days of end of treatment/study. | |
| Follow up/Survival Forms | Within 14 calendar days of the follow up activity. |
| Page 66 of 92 |
12.4 Regulatory Records
The investigator will maintain regulatory records, including updating records in accordance with Good Clinical Practice guidelines and FDA regulations
12.5 Protocol Deviations and Single Subject Exceptions
It is understood that deviations from the protocol should be avoided, except when necessary to eliminate an immediate hazard to a research participant. Brief interruptions and delays may occasionally be required because of travel delays, airport closures, inclement weather, family responsibilities, security alerts, government holidays, and so forth. Delays can also extend to complications of disease or unrelated medical illnesses not related to disease progression. The PI has the discretion to deviate from the protocol when necessary so long as such a deviation does not threaten patient safety or protocol scientific integrity. As a result of deviations, corrective actions are to be developed by the study staff and implemented promptly.
12.5.1 Definitions
| 12.5.1.1 | Deviation |
A deviation is a divergence from a specific element of a protocol that occurred without prior IRB approval. Investigators may deviate from the protocol to eliminate immediate hazard(s) for the protection, safety, and well-being of the study subjects without prior IRB approval. Examples include, but are not limited to: a) dose adjustments based on excessive patient weight; b) alteration in treatment schedule due to non-availability of the research participant for treatment; and c) laboratory test results which are slightly outside the protocol requirements but at levels that do not affect participant safety.
| 12.5.1.2 | Single Subject Exceptions (SSE) |
An SSE is a planned deviation, meaning that it involves circumstances in which the specific procedures called for in a protocol are not in the best interests of a specific patient. It is a deviation that is anticipated and receives prior approval by the Principal Investigator and the COH IRB.
12.5.2. Reporting of Deviations and SSEs
| 12.5.2.1 | Reporting Deviations |
For any deviation, the Investigator will notify the COH DSMC and IRB within 5 calendar days of its occurrence via iRIS in accordance with the Clinical Research Protocol Deviation policy.
2.2.2 Reporting Single Subject Exceptions
The SSE must be submitted as a “Single Subject Exception Amendment Request” via iRIS in accordance with IRB guidelines and the Clinical Research Protocol Deviation policy. An IRB approved SSE does not need to be submitted as a deviation to the DSMC.
In addition, if contractually obligated, the sponsor must also approve the deviation
| Page 67 of 92 |
| 13.0 |
Statistical
Considerations
|
13.1 Study Design
The Phase I dose-finding will use a traditional 3+3 to determine the MTD. These designs are based on first cycle DLTs. The determination of the RP2D will be based on the MTD (and will not exceed the MTD) and additional consideration of dose modifications, adverse events in subsequent cycles, clinical activity and correlative studies.
Safety: All patients who receive at least one dose of study drug will be evaluated for safety and toxicity. Safety analyses will include the following: summaries of the adverse event rates (including all events and study drug-related events), all serious adverse events (SAEs), deaths on-study, deaths within 30 days of the last dose of study drug, and discontinuations from study drug due to adverse events; listings and frequency tables categorizing laboratory and nonlaboratory adverse events by maximum CTCAE grade and relationship to study drug. Safety and efficacy will be reviewed at least every six months by COH DSMC.
Expanded Cohort: Expansion to 12 patients at the initial recommended Phase 2 dose (RP2D) will help confirm the choice of RP2D. In particular, if during the expansion cohort, more than 30% of the patients at the initial RP2D experience a DLT, the study will hold accrual (accrual can also be held at the discretion of the PI for non-DLT or other safety considerations). In addition, with 12 patients, any serious treatment-related adverse event that occurs with a true frequency of 10%, will be observed at least once with a probability of 72%, and any such AE with a true frequency of 20% would be observed at least once with a probability of 93%. The DLT rate can be estimated with a standard error of at most 14%.
Pharmacokinetic: PK analyses will be collected on all patients in the Phase I portion of the study. Exploratory graphical analysis will be conducted. PK/PD modeling may be conducted if deemed appropriate and necessary.
Patients will plan to receive treatment until progression, unacceptable toxicity or one of the criteria in 5.3 requires discontinuation. Each cycle is defined as 3 weeks (21 days). Patients who discontinue without disease progression will continue to be evaluated for tumor response using RECIST v1.1 (Appendix B) guidelines every 6-8 weeks until disease progression, death, or study closure.
| Page 68 of 92 |
Figure 1 illustrates the study design
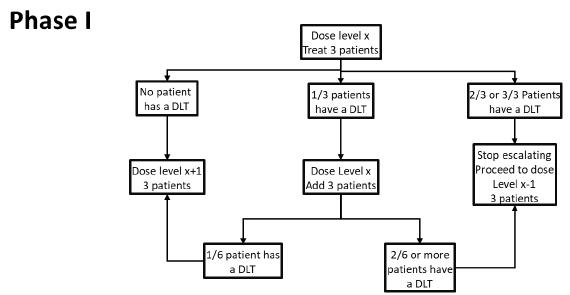
13.2 Sample Size Accrual Rate
The Phase Ib study accrual is expected to require 15 patients to determine the MTD (minimum 8, maximum 30, assuming the lowest dose is tolerated). With the expanded cohort, the expected sample-size is 21, and the maximum is 36 patients. This does not include patients who are not evaluable for DLT consideration during the dose finding. The study duration of the Phase Ib will be approximately 18-24 months.
13.3 Statistical Analysis Plan
| 13.3.1 | General Considerations |
Efficacy and safety analyses will include all randomized patients who received at least one dose of study drug. Patients will be grouped according to treatment received in Cycle 1. If a patient is randomized and it is confirmed later that they do not have SCLC then they will not be analyzed for efficacy but safety only.
Unless otherwise specified, missing data will not be imputed and kept missing in the data analysis. If patients miss a planned assessment, and have progression on the next assessment, the date of progression for purposes of analysis will be based on the planned assessment date that was missed.
| 13.3.2 | Patient Disposition |
A detailed description of patient disposition will be provided. It will include a summary of the number and percentage of patients entered into the study, enrolled in the study, and treated as well as number and percentage of patients completing the study, or discontinuing (overall and by reason for discontinuation). A summary of all important protocol violations will be provided.
| 13.3.3 | Patient Characteristics |
Patient demographics including age, gender, screening height and weight, and other demographic characteristics will be summarized using descriptive statistics.
Baseline disease characteristics will be summarized by presenting frequency counts and percentages for disease characteristics as appropriate.
| Page 69 of 92 |
| 13.3.4 | Concomitant Therapy |
Concomitant medications will be summarized for the safety population.
| 13.3.5 | Postdiscontinuation Therapy |
The numbers and percentages of patients reporting postdiscontinuation therapies will be provided overall, by type of therapy (surgery, radiotherapy, or systemic therapy), and by drug name.
| 13.3.6 | Treatment Compliance |
The number of dose omissions, reductions and delays will be summarized for all treated patients per treatment arm.
| 13.3.7 | Primary Outcome and Methodology |
The primary outcome measure for this study is to determine the recommended dose of LB-100 with carboplatin/etoposide/atezolizumab in patients with ED-SCLC.
| 13.3.8 | Pharmacokinetic/Pharmacodynamic Analyses |
Pharmacokinetics and Pharmacodynamic analyses will be conducted on all available LB-100 plasma concentration and pharmacodynamic data. These analyses are exploratory in nature. Graphical representation of the data will be conducted. PK/PD modeling may be pursued if deemed appropriate and necessary.
| 13.3.9 | Safety Analyses |
All safety summaries and analyses will be based upon the Safety Population as defined in Section 13.1.
Overall exposure to study drug, the numbers of patients completing each cycle, and the will be summarized using descriptive statistics.
An overall summary of AEs will be provided for AEs deemed by the investigator to be possibly related to study drug, and for all causalities.
A treatment-emergent adverse event (TEAE) is defined as an event that first occurred or worsened in severity after baseline.
The number of patients who experienced a TEAE, SAE, TEAE related to study drug, died, or discontinued from the study due to an AE will be summarized by treatment.
Common Terminology Criteria for Adverse Events v 5.0 will be used when reporting AEs by CTCAE terms. Laboratory and non-laboratory CTCAEs will be summarized by CTCAE term and maximum CTCAE grade, including the total for maximum Grade 3 and 4. These summaries will be provided for events regardless of study drug causality, and for events deemed by the investigator to be possibly related to study medication.
Reasons for death will be summarized separately for on-therapy and within 30 days of last dose of study drug. Serious adverse events will be summarized by PT.
Hospitalizations and transfusions during the study treatment period or during the 30-day follow-up period will be summarized by treatment group.
| 13.3.10 | Interim Analyses |
No formal interim analyses are planned for this study other than the safety assessments and scheduled external review.
| Page 70 of 92 |
| 14.0 | Human Subject Issues | |
| 14.1 | Institutional Review Board |
In accordance with City of Hope policies, an Institutional Review Board (IRB) that complies with the federal regulations at 45 CFR 46 and 21 CFR 50, 56 and State of California Health and Safety Code, Title 17, must review and approve this protocol and the informed consent form prior to initiation of the study. All institutional, NCI, Federal, and State of California regulations must be fulfilled.
14.2 Recruitment of Subjects
Patients will be recruited at City of Hope by their Medical Oncologist based on their diagnosis and eligibility criteria. A Clinical Trial Research Nurse will screen the patient for the trial and if they meet all eligibility criteria they will be enrolled into the study.
14.3 Advertisements
Advertisements to include print, media (radio, television, billboards), telephone scripts, lay summary to be posted on City of Hope’s public Clinical Trials On-LineSM website, etc., will be reviewed and approved by the IRB prior to their use to recruit potential study subjects.
14.4 Study location and Performance Sites
This study will be performed at COH.
14.5 Confidentiality
This research will be conducted in compliance with federal and state of California requirements relating to protected health information (PHI).
14.6 Financial Obligations and Compensation
The investigational drug, LB-100, will be provided free of charge by Lixte Biotechnology, Inc. East Setauket, NY 11133. Should this drug become commercially available during the course of your treatment, the research participant and/or the insurance carrier may be asked to pay for the costs of the drug.
The standard of care drugs and procedures provided will be the responsibility of the research participant and/or the insurance carrier. The research participant will be responsible for all copayments, deductibles, and other costs of treatment and diagnostic procedures as set forth by the insurance carrier. The research participant and/or the insurance carrier will be billed for the costs of treatment and diagnostic procedures in the same way as if the research participant were not in a research study. However, neither the research participant nor the insurance carrier will be responsible for the research procedures related to this study.
In the event of physical injury to a research participant, resulting from research procedures, appropriate medical treatment will be available at the City of Hope to the injured research participant, however, financial compensation will not be available.
The research participant will not be paid for taking part in this study.
| Page 71 of 92 |
14.7 Informed Consent Processes/Regulatory Considerations
The Principal Investigator or IRB approved named designate will explain the nature, duration, purpose of the study, potential risks, alternatives and potential benefits, and all other information contained in the informed consent document. In addition, they will review the experimental subject’s bill of rights and the HIPAA research authorization form. Research subjects will be informed that they may withdraw from the study at any time and for any reason without prejudice, including as applicable, their current or future care or employment at City of Hope or any relationship they have with City of Hope. Research subjects will be afforded sufficient time to consider whether or not to participate in the research.
Should sufficient doubt be raised regarding the adequacy of comprehension, further clarifications will be made and the questionnaire repeated until a satisfactory result is obtained. Prospective research subjects who cannot adequately comprehend the fundamental aspects of the research study with a reasonable amount of discussion, education and proctoring will be ineligible for enrollment. For those subjects who do comprehend the fundamental aspects of the study, consent will be obtained and documented, followed by eligibility testing. The research team will review the results of eligibility testing and determine if the subject is a candidate for study enrollment.
The Informed Consent Form (ICF) will be used to explain the potential risks and benefits of study participation to the patient in simple terms before the patient is entered into the study, and to document that the patient is satisfied with his or her understanding of the risks and benefits of participating in the study and desires to participate in the study.
The investigator is responsible for ensuring that informed consent is given by each patient or legal representative. This includes obtaining the appropriate signatures and dates on the ICF prior to the performance of any protocol procedures and prior to the administration of study drug.
As used in this protocol, the term “informed consent” includes all consent and assent given by patients or their legal representatives.
14.7.1 Regulatory Considerations
This study will be conducted in accordance with:
| 1) | Consensus ethics principles derived from international ethics guidelines, including the declaration of Helsinki and Council for International Organizations of Medical Sciences (CIOMS) International Ethical Guidelines | |
| 2) | The International Conference on Harmonisation (ICH) Good Clinical Practices (GCP) Guideline [E6] | |
| 3) | Applicable laws and regulations |
The investigator or designee will promptly submit the protocol to applicable ERB(s). LB-100 is being studied in the United States (US) under a US Investigational New Drug (IND) application. The US IND number is 151424.
All or some of the obligations of the sponsore will be assigned to a CRO.
An identification code assigned by the investigator for each patient will be used in lieu of the patient’s name to protect the patient’s identity when reporting AEs and/or other trial-related data.
| Page 72 of 92 |
14.7.2 Investigator Information
Site-specific contact information is provided in a separate document.
14.7.3 Protocol Signatures
After reading the protocol, each principal investigator will signt he protocol signature page and send a copy of the signed page to a City of Hope representative or designee.
14.7.4 Final Report Signature.
The sponsor’s responsible medical officer and responsible statistician will approve the final clinical study report for this study, confirming that, to the best of his or her knowledge, the report accurately describes the conduct and results of the study.
| 15.0 | Study Oversight, Quality Assurance, and Data & Safety Monitoring | |
| 15.1 | All Investigator Responsibilities |
An investigator is responsible for ensuring that an investigation is conducted according to the signed investigator statement, the investigational plan, and applicable regulations; for protecting the rights, safety, and welfare of subjects under the investigator’s care; and for the control of drugs under investigation.
All Investigators agree to:
| ● | Conduct the study in accordance with the protocol and only make changes after notifying the Sponsor (or designee), except when necessary to protect the safety, rights or welfare of subjects. | |
| ● | Personally conduct or supervise the study (or investigation). | |
| ● | Ensure that the requirements relating to obtaining informed consent and IRB review and approval meet federal guidelines, as stated in § 21 CFR, parts 50 and 56. | |
| ● | Report to the Sponsor or designee any AEs that occur in the course of the study, in accordance with §21 CFR 312.64. | |
| ● | Ensure that all associates, colleagues and employees assisting in the conduct of the study are informed about their obligations in meeting the above commitments. | |
| ● | Maintain adequate and accurate records in accordance with §21 CFR 312.62 and to make those records available for inspection with the Sponsor (or designee). | |
| ● | Ensure that an IRB that complies with the requirements of §21 CFR part 56 will be responsible for initial and continuing review and approval of the clinical study. | |
| ● | Promptly report to the IRB and the Sponsor all changes in the research activity and all unanticipated problems involving risks to subjects or others (to include amendments and IND safety reports). | |
| ● | Seek IRB and Sponsor approval before any changes are made in the research study, except when necessary to eliminate hazards to the patients/subjects. | |
| ● | Comply with all other requirements regarding the obligations of clinical investigators and all other pertinent requirements listed in § 21 CFR part 312. |
| Page 73 of 92 |
15.2 Study Principal Investigator Responsibilities
The Study Principal Investigator is responsible for the conduct of the clinical trial, including overseeing that sponsor responsibilities as defined in § 21 CFR 312. Subpart D are executed in accordance with federal regulations.
15.3 Protocol Management Team (PMT)
The Protocol Management Team (PMT), minimally consisting of the study PI, collaborating investigators, research nurse, clinical research associate/coordinator, and the study biostatistician, is responsible for ongoing monitoring of the data and safety of this study, including implementation of the stopping rules for safety/toxicity.
The PMT is recommended to meet (in person or via teleconference) at least monthly to review study status. This review will include, but not be limited to, reportable AEs and UPs, and an update of the ongoing study summary that describes study progress in terms of the study schema. The meeting will be a forum to discuss study related issues including accrual, SAE/AEs experienced, study response, deviations/violations and study management issues. The appropriateness of further subject enrollment and the specific intervention for subsequent subject enrollment are addressed. It is recommended that minutes of these discussions be taken to document the date of these meetings, attendees and the issues that were discussed (in a general format).
15.4 Monitoring
Clinical site monitoring is conducted to ensure that the rights of human subjects are protected, that the study is implemented in accordance with the protocol and regulatory requirements, and that the quality and integrity of study data and data collection methods are maintained. Monitoring for this study will be performed by the City of Hope Office of Clinical Trials Auditing and Monitoring (OCTAM).
The Investigator will permit the study monitors and appropriate regulatory authorities direct access to the study data and to the corresponding source data and documents to verify the accuracy of this data. The Investigator will allocate adequate time for such monitoring activities. The Investigator will also ensure that the monitor or other compliance or quality assurance reviewer is given access to all the above noted study-related documents and study related facilities (e.g. pharmacy, diagnostic laboratory, etc.), and has adequate space to conduct the monitoring visit.
Details of clinical site monitoring are documented in the OCTAM SOP. This document specifies the frequency of monitoring, monitoring procedures, the level of clinical site monitoring activities (e.g., the percentage of subject data to be reviewed), and the distribution of monitoring reports. Staff from OCTAM will conduct monitoring activities and provide reports of the findings and associated action items in accordance with the details described in the SOP. Documentation of monitoring activities and findings will be provided to the study team, and the COH DSMC.
15.5 Quality Assurance
The City of Hope Clinical Research Information Support will provide support for this trial as detailed in the COH DCC Operations Plan provided as a supplement to this document.
| Page 74 of 92 |
Quality assurance (QA) audits will be conducted by Theradex Oncology. The QA audits will be conducted on an annual schedule with the first QA audit conducted approximately 6 months after initiation of patient enrollment. QA audits will be conducted similar to those conducted on behalf of the NCI Experimental Therapeutics Clinical Trial Network (ETCTN) guidelines: https://ctep.cancer.gov/branches/ctmb/clinicalTrials/docs/ETCTN_Audit_Guidelines.pdf
15.6 City of Hope Data and Safety Monitoring Committee
This is a risk level4 study as defined in the City of Hope Institutional Data and Safety Monitoring Plan. This determination was made because the study involves a COH held IND.
The DSMC is a multidisciplinary committee charged with overseeing the monitoring of safety of participants in clinical trials, and the conduct, progress, validity, and integrity of the data for all clinical trials that are sponsored by City of Hope. The committee is composed of clinical specialists with experience in oncology and who have no direct relationship with the study. The committee reviews the progress and safety of all active research protocols that are not monitored by another safety and data monitoring committee or board.
The Study Principal Investigator is required to submit periodic status reports (the PMT report) according to the guidelines outlined in the City of Hope Institutional Data and Safety Monitoring Plan. The PMT report will be submitted to the COH DSMC quarterly from the date of activation.
The COH Data and Safety Monitoring Committee (DSMC) will review and monitor toxicity and accrual data from this trial. The DSMC will review up-to-date participant accrual; summary of all adverse events captured via routine and expedited reporting; a summary of deviations; any response information; monitoring reports, and summary comments provided by the study team. Other information (e.g. scans, laboratory values) will be provided upon request. For Phase I studies, a Phase I Tracking Log will be utilized and reviewed by the DSMC to monitor data and safety for dose escalation. A review of outcome results (response, toxicity and adverse events) and factors external to the study (such as scientific or therapeutic developments) is discussed, and the Committee votes on the status of each study. Information that raises any questions about participant safety will be addressed with the Principal Investigator, statistician and study team.
| Page 75 of 92 |
| VII. | 16.0 References |
1. Quoix E, Breton JL, Daniel C, Jacoulet P, Debieuvre D, Paillot N, Kessler R, Moreau L, Coetmeur D, Lemarie E, Milleron B. Etoposide phosphate with carboplatin in the treatment of elderly patients with small-cell lung cancer: a phase II study. Ann Oncol. 2001;12(7):957-62. Epub 2001/08/28. PubMed PMID: 11521802.
2. Maghfoor I, Perry MC. Lung cancer. Ann Saudi Med. 2005;25(1):1-12. Epub 2005/04/13. PubMed PMID: 15822487; PMCID: PMC6150570.
3. Niell HB, Perry MC, Clamon G, Crawford J, Miller AA, Herndon J, 2nd, Green MR. Carboplatin/etoposide/paclitaxel in the treatment of patients with extensive small-cell lung cancer. Clin Lung Cancer. 2001;2(3):204-9. Epub 2004/01/01. PubMed PMID: 14700479.
4. Sundstrom S, Bremnes RM, Kaasa S, Aasebo U, Hatlevoll R, Dahle R, Boye N, Wang M, Vigander T, Vilsvik J, Skovlund E, Hannisdal E, Aamdal S, Norwegian Lung Cancer Study G. Cisplatin and etoposide regimen is superior to cyclophosphamide, epirubicin, and vincristine regimen in small-cell lung cancer: results from a randomized phase III trial with 5 years’ follow-up. J Clin Oncol. 2002;20(24):4665-72. Epub 2002/12/19. doi: 10.1200/JCO.2002.12.111. PubMed PMID: 12488411.
5. Niell HB, Herndon JE, 2nd, Miller AA, Watson DM, Sandler AB, Kelly K, Marks RS, Perry MC, Ansari RH, Otterson G, Ellerton J, Vokes EE, Green MR, Cancer, Leukemia G. Randomized phase III intergroup trial of etoposide and cisplatin with or without paclitaxel and granulocyte colony-stimulating factor in patients with extensive-stage small-cell lung cancer: Cancer and Leukemia Group B Trial 9732. J Clin Oncol. 2005;23(16):3752-9. doi: 10.1200/JCO.2005.09.071. PubMed PMID: 15923572.
6. Eckardt JR, von Pawel J, Papai Z, Tomova A, Tzekova V, Crofts TE, Brannon S, Wissel P, Ross G. Open-label, multicenter, randomized, phase III study comparing oral topotecan/cisplatin versus etoposide/cisplatin as treatment for chemotherapy-naive patients with extensive-disease small-cell lung cancer. J Clin Oncol. 2006;24(13):2044-51. Epub 2006/05/02. doi: 10.1200/JCO.2005.03.3332. PubMed PMID: 16648504.
7. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M, Reck M, Mok T, Lam S, Shames DS, Liu J, Ding B, Lopez-Chavez A, Kabbinavar F, Lin W, Sandler A, Liu SV, Group IMS. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med. 2018;379(23):2220- 9. doi: 10.1056/NEJMoa1809064. PubMed PMID: 30280641.
8. Bunn PA, Jr. Review of therapeutic trials of carboplatin in lung cancer. Semin Oncol. 1989;16(2 Suppl 5):27-33. Epub 1989/04/01. PubMed PMID: 2541506.
| Page 76 of 92 |
9. Bunn PA, Jr., Kelly K. A phase I study of carboplatin and paclitaxel in non-small cell lung cancer: a University of Colorado Cancer Center study. Semin Oncol. 1995;22(4 Suppl 9):2-6. Epub 1995/08/01. PubMed PMID: 7644924.
10. Prendiville J, Lorigan P, Hicks F, Leahy B, Stout R, Burt P, Thatcher N. Therapy for small cell lung cancer using carboplatin, ifosfamide, etoposide (without dose reduction), mid-cycle vincristine with thoracic and cranial irradiation. Eur J Cancer. 1994;30A(14):2085- 90. Epub 1994/01/01. PubMed PMID: 7857708.
11. Gatzemeier U, Hossfeld DK, Neuhauss R, Reck M, Achterrath W, Lenaz L. Phase II and III studies with carboplatin in small cell lung cancer. Semin Oncol. 1992;19(1 Suppl 2):28-36. Epub 1992/02/01. PubMed PMID: 1329220.
12. Larive S, Bombaron P, Riou R, Fournel P, Perol M, Lena H, Dussopt C, Philip-Joet F, Touraine F, Lecaer H, Souquet PJ, Groupe Lyon-Saint Etienne d’Oncologie T. Carboplatin-etoposide combination in small cell lung cancer patients older than 70 years: a phase II trial. Lung Cancer. 2002;35(1):1-7. Epub 2001/12/26. PubMed PMID: 11750705.
13. Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 2013;14(6):e229-38. Epub 2013/05/04. doi: 10.1016/S1470-2045(12)70558-2. PubMed PMID: 23639323; PMCID: PMC3913484.
14. Hong CS, Ho W, Zhang C, Yang C, Elder JB, Zhuang Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol Ther. 2015;16(6):821-33. Epub 2015/04/22. doi: 10.1080/15384047.2015.1040961. PubMed PMID: 25897893; PMCID: PMC4623051.
15. Chung V, Mansfield AS, Braiteh F, Richards D, Durivage H, Ungerleider RS, Johnson F, Kovach JS. Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin Cancer Res. 2017;23(13):3277-84. Epub 2017/01/01. doi: 10.1158/1078-0432.CCR-16-2299. PubMed PMID: 28039265.
16. Lu J, Kovach JS, Johnson F, Chiang J, Hodes R, Lonser R, Zhuang Z. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc Natl Acad Sci U S A. 2009;106(28):11697-702. Epub 2009/07/01. doi: 10.1073/pnas.0905930106. PubMed PMID: 19564615; PMCID: PMC2710674.
17. Wang Y, Yang R, Gu J, Yin X, Jin N, Xie S, Wang Y, Chang H, Qian W, Shi J, Iqbal K, Gong CX, Cheng C, Liu F. Cross talk between PI3K-AKT- GSK-3beta and PP2A pathways determines tau hyperphosphorylation. Neurobiol Aging. 2015;36(1):188-200. Epub 2014/09/16. doi: 10.1016/j.neurobiolaging.2014.07.035. PubMed PMID: 25219467.
18. Liu GP, Wei W, Zhou X, Shi HR, Liu XH, Chai GS, Yao XQ, Zhang JY, Peng CX, Hu J, Li XC, Wang Q, Wang JZ. Silencing PP2A inhibitor by lenti-shRNA interference ameliorates neuropathologies and memory deficits in tg2576 mice. Mol Ther. 2013;21(12):2247-57. Epub 2013/08/08. doi: 10.1038/mt.2013.189. PubMed PMID: 23922015; PMCID: PMC3863796.
| Page 77 of 92 |
19. Gordon IK, Lu J, Graves CA, Huntoon K, Frerich JM, Hanson RH, Wang X, Hong CS, Ho W, Feldman MJ, Ikejiri B, Bisht K, Chen XS, Tandle A, Yang C, Arscott WT, Ye D, Heiss JD, Lonser RR, Camphausen K, Zhuang Z. Protein Phosphatase 2A Inhibition with LB100 Enhances Radiation-Induced Mitotic Catastrophe and Tumor Growth Delay in Glioblastoma. Mol Cancer Ther. 2015;14(7):1540-7. Epub 2015/05/06. doi: 10.1158/1535-7163.MCT- 14-0614. PubMed PMID: 25939762; PMCID: PMC4497833.
20. Zhu XN, Chen LP, Bai Q, Ma L, Li DC, Zhang JM, Gao C, Lei ZN, Zhang ZB, Xing XM, Liu CX, He ZN, Li J, Xiao YM, Zhang AH, Zeng XW, Chen W. PP2A-AMPKalpha-HSF1 axis regulates the metal-inducible expression of HSPs and ROS clearance. Cell Signal. 2014;26(4):825-32. Epub 2014/01/15. doi: 10.1016/j.cellsig.2014.01.002. PubMed PMID: 24412756.
21. Xiao G, Chan LN, Klemm L, Braas D, Chen Z, Geng H, Zhang QC, Aghajanirefah A, Cosgun KN, Sadras T, Lee J, Mirzapoiazova T, Salgia R, Ernst T, Hochhaus A, Jumaa H, Jiang X, Weinstock DM, Graeber TG, Muschen M. B-Cell-Specific Diversion of Glucose Carbon Utilization Reveals a Unique Vulnerability in B Cell Malignancies. Cell. 2018;173(2):470-84 e18. Epub 2018/03/20. doi: 10.1016/j.cell.2018.02.048. PubMed PMID: 29551267; PMCID: PMC6284818.
22. Zhuang Z, Lu J, Lonser R, Kovach JS. Enhancement of cancer chemotherapy by simultaneously altering cell cycle progression and DNA- damage defenses through global modification of the serine/threonine phospho-proteome. Cell Cycle. 2009;8(20):3303-6. Epub 2009/10/07. doi: 10.4161/cc.8.20.9689. PubMed PMID: 19806030.
23. Lu J, Zhuang Z, Song DK, Mehta GU, Ikejiri B, Mushlin H, Park DM, Lonser RR. The effect of a PP2A inhibitor on the nuclear receptor corepressor pathway in glioma. J Neurosurg. 2010;113(2):225-33. Epub 2009/12/17. doi: 10.3171/2009.11.JNS091272. PubMed PMID: 20001590.
24. Martiniova L, Lu J, Chiang J, Bernardo M, Lonser R, Zhuang Z, Pacak K. Pharmacologic modulation of serine/threonine phosphorylation highly sensitizes PHEO in a MPC cell and mouse model to conventional chemotherapy. PLoS One. 2011;6(2):e14678. Epub 2011/02/23. doi: 10.1371/journal.pone.0014678. PubMed PMID: 21339823; PMCID: PMC3038858.
25. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, Joseph RW, Galsky MD, Fleming MT, Petrylak DP, Perez-Gracia JL, Burris HA, Castellano D, Canil C, Bellmunt J, Bajorin D, Nickles D, Bourgon R, Frampton GM, Cui N, Mariathasan S, Abidoye O, Fine GD, Dreicer R. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909-20. Epub 2016/03/10. doi: 10.1016/S0140-6736(16)00561-4. PubMed PMID: 26952546; PMCID: PMC5480242.
| Page 78 of 92 |
26. Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, Loriot Y, Necchi A, Hoffman-Censits J, Perez-Gracia JL, Dawson NA, van der Heijden MS, Dreicer R, Srinivas S, Retz MM, Joseph RW, Drakaki A, Vaishampayan UN, Sridhar SS, Quinn DI, Duran I, Shaffer DR, Eigl BJ, Grivas PD, Yu EY, Li S, Kadel EE, 3rd, Boyd Z, Bourgon R, Hegde PS, Mariathasan S, Thastrom A, Abidoye OO, Fine GD, Bajorin DF, Group IMS. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389(10064):67-76. Epub 2016/12/13. doi: 10.1016/S0140-6736(16)32455-2. PubMed PMID: 27939400; PMCID: PMC5568632.
27. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, Braiteh F, Waterkamp D, He P, Zou W, Chen DS, Yi J, Sandler A, Rittmeyer A, Group PS. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837-46. Epub 2016/03/14. doi: 10.1016/S0140-6736(16)00587-0. PubMed PMID: 26970723.
28. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA, Investigators IMT. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379(22):2108-21. Epub 2018/10/23. doi: 10.1056/NEJMoa1809615. PubMed PMID: 30345906.
| Page 79 of 92 |
| (i) | APPENDIX A: PERFORMANCE STATUS SCALES |
| Karnofsky Scale % | Karnofsky Description |
ECOG Scale* |
ECOG Description |
|||
| 100 | Normal, no complaints, no evidence of disease. | 0 | Fully active, able to carry on all pre-disease activities without restriction | |||
| 90 | Able to carry on normal activity, minor symptoms or signs of disease. | 1 | Restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature e.g. light house work, office work. | |||
| 80 | Normal activity with effort, some signs or symptoms of disease. | |||||
| 70 | Cares for self, unable to carry on normal activity or to do active work. | 2 | Ambulatory and capable of all self care but unable to carry out any work activities. Up and about more than 50% of waking hours | |||
| 60 | Requires occasional assistance, but is able to care for most of own needs. | |||||
| 50 | Requires considerable assistance and frequent medical care. | 3 | Capable of only limited self care, confined to bed or chair more than 50% of waking hours | |||
| 40 | Disabled, requires special care and assistance. | |||||
| 30 | Severely disabled, hospitalization is indicated although death is not imminent. | 4 | Completely disabled. Cannot carry on any self care. Totally confined to bed or chair | |||
| 20 | Hospitalization necessary, very sick, active supportive treatment necessary. | |||||
| 10 | Moribund, fatal processes | |||||
| Dead | 5 | Dead |
*also known as Zubrod, SWOG or WHO scale
| Page 80 of 92 |
| (ii) | APPENDIX B: RECIST V1.1 CRITERIA |
1.1 Antitumor Effect – RECIST V1.1 Solid Tumors
Response and progression will be evaluated in this study using the new international criteria proposed by the revised Response Evaluation Criteria in Solid Tumors (RECIST) guideline (version 1.1) [Eur J Ca 45:228-247, 2009]. Changes in the largest diameter (unidimensional measurement) of the tumor lesions and the shortest diameter in the case of malignant lymph nodes are used in the RECIST criteria.
1.1.1 Definitions
Evaluable for Toxicity. All patients will be evaluable for toxicity from the time of their first treatment.
Evaluable for Objective Response. Only those patients who have measurable disease present at baseline, have received at least one cycle of therapy, and have had their disease re-evaluated will be considered evaluable for response. These patients will have their response classified according to the definitions stated below. (Note: Patients who exhibit objective disease progression prior to the end of cycle 1 will also be considered evaluable.)
Evaluable Non-Target Disease Response. Patients who have lesions present at baseline that are evaluable but do not meet the definitions of measurable disease, have received at least one cycle of therapy, and have had their disease re-evaluated will be considered evaluable for non-target disease. The response assessment is based on the presence, absence, or unequivocal progression of the lesions.
1.1.2 Disease Parameters
Measurable Disease. Measurable lesions are defined as those that can be accurately measured in at least one dimension (longest diameter to be recorded) as ≥20 mm (≥2 cm) by chest x-ray or as ≥10 mm (≥1 cm) with CT scan, MRI, or calipers by clinical exam. All tumor measurements must be recorded in millimeters (or decimal fractions of centimeters).
Note: Tumor lesions that are situated in a previously irradiated area might or might not be considered measurable.
Malignant Lymph Nodes. To be considered pathologically enlarged and measurable, a lymph node must be ≥15 mm (≥1.5 cm) in short axis when assessed by CT scan (CT scan slice thickness recommended to be no greater than 5 mm [0.5 cm]). At baseline and in follow-up, only the short axis will be measured and followed.
| Page 81 of 92 |
Non-Measurable Disease. All other lesions (or sites of disease), including small lesions (longest diameter <10 mm [<1 cm] or pathological lymph nodes with ≥10 to <15 mm [≥1 to <1.5 cm] short axis), are considered non-measurable disease. Bone lesions, leptomeningeal disease, ascites, pleural/pericardial effusions, lymphangitis cutis/pulmonitis, inflammatory breast disease, and abdominal masses (not followed by CT or MRI), are considered as non-measurable.
Note: Cystic lesions that meet the criteria for radiographically defined simple cysts should not be considered as malignant lesions (neither measurable nor non-measurable) since they are, by definition, simple cysts.
‘Cystic lesions’ thought to represent cystic metastases can be considered as measurable lesions, if they meet the definition of measurability described above. However, if non-cystic lesions are present in the same patient, these are preferred for selection as target lesions.
Target Lesions. All measurable lesions up to a maximum of 2 lesions per organ and 5 lesions in total, representative of all involved organs, should be identified as target lesions and recorded and measured at baseline. Target lesions should be selected on the basis of their size (lesions with the longest diameter), be representative of all involved organs, but in addition should be those that lend themselves to reproducible repeated measurements. It may be the case that, on occasion, the largest lesion does not lend itself to reproducible measurement in which circumstance the next largest lesion which can be measured reproducibly should be selected. A sum of the diameters (longest for non-nodal lesions, short axis for nodal lesions) for all target lesions will be calculated and reported as the baseline sum diameters. If lymph nodes are to be included in the sum, then only the short axis is added into the sum. The baseline sum diameters will be used as reference to further characterize any objective tumor regression in the measurable dimension of the disease.
Non-Target Lesions. All other lesions (or sites of disease) including any measurable lesions over and above the 5 target lesions should be identified as non-target lesions and should also be recorded at baseline. Measurements of these lesions are not required, but the presence, absence, or in rare cases unequivocal progression of each should be noted throughout follow-up.
1.1.3 Methods for Evaluation of Measurable Disease
All measurements should be taken and recorded in metric notation using a ruler or calipers. All baseline evaluations should be performed as closely as possible to the beginning of treatment and never more than 4 weeks before the beginning of the treatment.
| Page 82 of 92 |
The same method of assessment and the same technique should be used to characterize each identified and reported lesion at baseline and during follow-up. Imaging-based evaluation is preferred to evaluation by clinical examination unless the lesion(s) being followed cannot be imaged but are assessable by clinical exam.
Clinical Lesions. Clinical lesions will only be considered measurable when they are superficial (e.g., skin nodules and palpable lymph nodes) and ≥10 mm (≥1 cm) diameter as assessed using calipers (e.g., skin nodules). In the case of skin lesions, documentation by color photography, including a ruler to estimate the size of the lesion, is recommended.
Chest X-Ray. Lesions on chest x-ray are acceptable as measurable lesions when they are clearly defined and surrounded by aerated lung. However, CT is preferable.
Conventional CT and MRI. This guideline has defined measurability of lesions on CT scan based on the assumption that CT slice thickness is 5 mm (0.5 cm) or less. If CT scans have slice thickness greater than 5 mm (0.5 cm), the minimum size for a measurable lesion should be twice the slice thickness. MRI is also acceptable in certain situations (e.g. for body scans).
Use of MRI remains a complex issue. MRI has excellent contrast, spatial, and temporal resolution; however, there are many image acquisition variables involved in MRI, which greatly impact image quality, lesion conspicuity, and measurement. Furthermore, the availability of MRI is variable globally. As with CT, if an MRI is performed, the technical specifications of the scanning sequences used should be optimized for the evaluation of the type and site of disease. Furthermore, as with CT, the modality used at follow-up should be the same as was used at baseline and the lesions should be measured/assessed on the same pulse sequence. It is beyond the scope of the RECIST guidelines to prescribe specific MRI pulse sequence parameters for all scanners, body parts, and diseases. Ideally, the same type of scanner should be used and the image acquisition protocol should be followed as closely as possible to prior scans. Body scans should be performed with breath-hold scanning techniques, if possible.
PET-CT. At present, the low dose or attenuation correction CT portion of a combined PET-CT is not always of optimal diagnostic CT quality for use with RECIST measurements. However, if the site can document that the CT performed as part of a PET-CT is of identical diagnostic quality to a diagnostic CT (with IV and oral contrast), then the CT portion of the PET-CT can be used for RECIST measurements and can be used interchangeably with conventional CT in accurately measuring cancer lesions over time. Note, however, that the PET portion of the CT introduces additional data which may bias an investigator if it is not routinely or serially performed.
Ultrasound. Ultrasound is not useful in assessment of lesion size and should not be used as a method of measurement. Ultrasound examinations cannot be reproduced in their entirety for independent review at a later date and, because they are operator dependent, it cannot be guaranteed that the same technique and measurements will be taken from one assessment to the next. If new lesions are identified by ultrasound in the course of the study, confirmation by CT or MRI is advised. If there is concern about radiation exposure at CT, MRI may be used instead of CT in selected instances.
| Page 83 of 92 |
Endoscopy, Laparoscopy. The utilization of these techniques for objective tumor evaluation is not advised. However, such techniques may be useful to confirm complete pathological response when biopsies are obtained or to determine relapse in trials where recurrence following complete response (CR) or surgical resection is an endpoint.
Tumor Markers. Tumor markers alone cannot be used to assess response. If markers are initially above the upper normal limit, they must normalize for a patient to be considered in complete clinical response. Specific guidelines for both CA-125 response (in recurrent ovarian cancer) and PSA response (in recurrent prostate cancer) have been published [JNCI 96:487-488, 2004; J Clin Oncol 17, 3461-3467, 1999; J Clin Oncol 26:1148-1159, 2008]. In addition, the Gynecologic Cancer Intergroup has developed CA-125 progression criteria which are to be integrated with objective tumor assessment for use in first-line trials in ovarian cancer [JNCI 92:1534-1535, 2000].
Cytology, Histology. These techniques can be used to differentiate between partial responses (PR) and complete responses (CR) in rare cases (e.g., residual lesions in tumor types, such as germ cell tumors, where known residual benign tumors can remain).
The cytological confirmation of the neoplastic origin of any effusion that appears or worsens during treatment when the measurable tumor has met criteria for response or stable disease is mandatory to differentiate between response or stable disease (an effusion may be a side effect of the treatment) and progressive disease.
FDG-PET. While FDG-PET response assessments need additional study, it is sometimes reasonable to incorporate the use of FDG-PET scanning to complement CT scanning in assessment of progression (particularly possible ‘new’ disease). New lesions on the basis of FDG-PET imaging can be identified according to the following algorithm:
| a. | Negative FDG-PET at baseline, with a positive FDG-PET at follow-up is a sign of PD based on a new lesion. | |
| b. | No FDG-PET at baseline and a positive FDG-PET at follow-up: If the positive FDG-PET at follow-up corresponds to a new site of disease confirmed by CT, this is PD. If the positive FDG- PET at follow-up is not confirmed as a new site of disease on CT, additional follow-up CT scans are needed to determine if there is truly progression occurring at that site (if so, the date of PD will be the date of the initial abnormal FDG-PET scan). If the positive FDG-PET at follow-up corresponds to a pre-existing site of disease on CT that is not progressing on the basis of the anatomic images, this is not PD. | |
| c. |
FDG-PET may be used to upgrade a response to a CR in a manner similar to a biopsy in cases where a residual radiographic abnormality is thought to represent fibrosis or scarring. The use of FDG-PET in this circumstance should be prospectively described in the protocol and supported by disease-specific medical literature for the indication. However, it must be acknowledged that both approaches may lead to false positive CR due to limitations of FDG-PET and biopsy resolution/sensitivity. |
| Page 84 of 92 |
Note: A ‘positive’ FDG-PET scan lesion means one which is FDG avid with an uptake greater than twice that of the surrounding tissue on the attenuation corrected image.
1.1.4 Response Criteria
Evaluation of Target Lesions
Complete Response (CR): Disappearance of all target lesions. Any pathological lymph nodes (whether target or non-target) must have reduction in short axis to <10 mm (<1 cm).
Partial Response (PR): At least a 30% decrease in the sum of the diameters of target lesions, taking as reference the baseline sum diameters.
Progressive Disease (PD): At least a 20% increase in the sum of the diameters of target lesions, taking as reference the smallest sum on study (this includes the baseline sum if that is the smallest on study). In addition to the relative increase of 20%, the sum must also demonstrate an absolute increase of at least 5 mm (0.5 cm). (Note: the appearance of one or more new lesions is also considered progressions).
Stable Disease (SD): Neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD, taking as reference the smallest sum diameters while on study.
(b) Evaluation of Non-Target Lesions
Complete Response (CR): Disappearance of all non-target lesions and normalization of tumor marker level. All lymph nodes must be non-pathological in size (<10 mm [<1 cm] short axis).
Note: If tumor markers are initially above the upper normal limit, they must normalize for a patient to be considered in complete clinical response.
Non-CR/Non-PD: Persistence of one or more non-target lesion(s) and/or maintenance of tumor marker level above the normal limits.
| Page 85 of 92 |
Progressive Disease (PD): Appearance of one or more new lesions and/or unequivocal progression of existing non-target lesions. Unequivocal progression should not normally trump target lesion status. It must be representative of overall disease status change, not a single lesion increase.
Although a clear progression of “non-target” lesions only is exceptional, the opinion of the treating physician should prevail in such circumstances, and the progression status should be confirmed at a later time by the review panel (or Principal Investigator).
(c) Evaluation of Best Overall Response
The best overall response is the best response recorded from the start of the treatment until disease progression/recurrence (taking as reference for progressive disease the smallest measurements recorded since the treatment started). The patient’s best response assignment will depend on the achievement of both measurement and confirmation criteria.
(i) For Patients with Measurable Disease (i.e., Target Disease)
| Target Lesions |
Non-Target Lesions |
New Lesions | Overall Response |
Best Overall Response when Confirmation is Required* |
||||
| CR | CR | No | CR | ≥4 wks. Confirmation** | ||||
| CR | Non-CR/Non-PD | No | PR | |||||
| CR | Not evaluated | No | PR | ≥4 wks. Confirmation** | ||||
| PR | Non-CR/Non- PD/not evaluated | No | PR | |||||
| SD | Non-CR/Non- PD/not evaluated | No | SD | Documented at least once ≥4 wks. from baseline** | ||||
| PD | Any | Yes or No | PD | |||||
| Any | PD*** | Yes or No | PD | no prior SD, PR or CR | ||||
| Any | Any | Yes | PD |
| * | See RECIST 1.1 manuscript for further details on what is evidence of a new lesion. |
| ** | Only for non-randomized trials with response as primary endpoint. |
| *** | In exceptional circumstances, unequivocal progression in non-target lesions may be accepted as disease progression. |
| Note: | Patients with a global deterioration of health status requiring discontinuation of treatment without objective evidence of disease progression at that time should be reported as “symptomatic deterioration.” Every effort should be made to document the objective progression even after discontinuation of treatment. |
| Page 86 of 92 |
For Patients with Non-Measurable Disease (i.e., Non-Target Disease)
| Non-Target Lesions | New Lesions | Overall Response | ||
| CR | No | CR | ||
| Non-CR/non-PD | No | Non-CR/non-PD* | ||
| Not all evaluated | No | not evaluated | ||
| Unequivocal PD | Yes or No | PD | ||
| Any | Yes | PD |
| * | ‘Non-CR/non-PD’ is preferred over ‘stable disease’ for non-target disease since SD is increasingly used as an endpoint for assessment of efficacy in some trials so to assign this category when no lesions can be measured is not advised |
1.1.5 Duration of Response
Duration of overall response: The duration of overall response is measured from the time measurement criteria are met for CR or PR (whichever is first recorded) until the first date that recurrent or progressive disease is objectively documented (taking as reference for progressive disease the smallest measurements recorded since the treatment started).
The duration of overall CR is measured from the time measurement criteria are first met for CR until the first date that progressive disease is objectively documented.
Duration of stable disease: Stable disease is measured from the start of the treatment until the criteria for progression are met, taking as reference the smallest measurements recorded since the treatment started, including the baseline measurements.
1.1.6 Progression-Free Survival
PFS is defined as the duration of time from start of treatment to time of progression or death, whichever occurs first.
| Page 87 of 92 |
| (ii) | APPENDIX C: REGISTRATION COVERSHEET |
COH IRB# 20068: A PHASE Ib OPEN-LABEL STUDY OF LB-100 IN COMBINATION WITH
CARBOPLATIN/ETOPOSIDE/ATEZOLIZUMAB IN UNTREATED EXTENSIVE-STAGE
SMALL CELL LUNG CARCINOMA
| Data Coordinating Center: | Site Principal Investigator | |
|
City of Hope 1500 Duarte Road Duarte, CA 91010 Tel: (626)-218-7904 Email: DCC@coh.org (use #secure# in subject line) |
Name: Ravi Salgia, MD, PhD |
| CRA/Study Coordinator: | Contact Number: | |||||
| Patient’s Initials: (F M L): | Institution: | |||||
| PI/ Sub-Investigator: | ||||||
| Patient’s DOB: | IRB approval valid until (date): | |||||
| Sex: ____Male ____Female | Date Informed Consent Signed: | |||||
| Projected start date of treatment: | ||||||
| Race | Ethnicity | Method of Payment: _____ | ||||
| [ ] | Black | [ ] | Hispanic | Codes: | ||
| [ ] | Caucasian | [ ] | Non-Hispanic | 01 Private |
06 Military or Veterans Adm. sponsored |
|
| [ ] | Asian | [ ] | Other | 02 Medicare |
07 Self-pay (no insurance) |
|
| [ ] | American Indian | 03 Medicare & private ins. |
08 No means of payment (no insurance) |
|||
| [ ] | Native Hawaiian/Pacific Islander | 04 Medicaid | 09 Unknown | |||
| [ ] | Other ____ | 05 Medicaid & Medicare | ||||
Reason for Screen Failure:
Reason for Failing to Initiate Protocol Therapy:
| Page 88 of 92 |
| (iii) | APPENDIX D: EXPEDITED REPORTING COVERSHEET |
NOTIFICATION OF UNANTICIPATED PROBLEM/SERIOUS ADVERSE EVENT
THIS FORM ALONG WITH A COPY OF THE MEDWATCH 3500 OR IRB REPORTING FORM MUST BE EMAILED TO DCC@COH.ORG WITHIN 24 HOURS OF KNOWLEDGE OF ONSET OF SERIOUS ADVERSE EVENT OR UNANCTICIPATED PROBLEM
(iv) COH IRB # 20068
| From: | Date: |
| Phone No.: | Email: |
| Reporting Investigator: | |
| Event: | |
| Participant ID: | Institution: |
| Date Event Met Reporting Criteria (as defined in protocol): |
| Type of Report: | [ ] Initial [ ] Follow-up |
| CTCAE Grade: | [ ] G1/mild [ ] G2/moderate [ ] G3/severe [ ] G4/life threatening [ ] G5 |
| Attribution to Agent xx: | [ ] Not Applicable* [ ] Unrelated [ ] Unlikely [ ] Possible [ ] Probable [ ] Definite |
| Attribution to Agent xxy: | [ ] Not Applicable* [ ] Unrelated [ ] Unlikely [ ] Possible [ ] Probable [ ] Definite |
| Historical/Known Correlation to Agent xx: |
[ ] Expected [ ] Unexpected |
| Historical/Known Correlation to Agent xxy: |
[ ] Expected [ ] Unexpected |
| Meets Definition of Serious AE: | [ ] Serious [ ] Non-serious |
| Meets Definition of Unanticipated Problem: |
[ ] UP [ ] Not a UP |
| Has the event been reported to the following institution’s IRB? |
[ ] No [ ] Yes; Date:___/___/_____ |
* Not Applicable should only be used if subject has not received this agent.
|
Authorized Investigator Signature: |
Date:____/____/_____ |
| Page 89 of 92 |
| (v) | APPENDIX E: VETERANS ADMINISTRATION LUNG STUDY GROUP (VALG) STAGING SYSTEM FOR SCLC |
Veterans
Administration Lung Study
Group (VALG) Staging System for SCLC
| Stage | Characteristics | |
| Limited SCLC | Disease confined to the ipsilateral hemithorax which can be safely encompassed within a radiation field | |
| Extensive SCLC | Disease beyond the ipsilateral hemithorax, including malignant pleural or pericardial effusion or hematogenous metastases | |
SCLC = small cell lung cancer
| Page 90 of 92 |
APPENDIX F: COMMON SUBSTRATES OF CYP450 ISOENZYMES
These are only examples, please consult an updated source, such as the FDA website for the latest and most accurate information. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development- and-drug-interactions-table-substrates-inhibitors-and-inducers#classSub
| CYP3A group (includes 4, 5, and 7) |
Amiodarone Amlodipine Aripiprazole Atorvastatin Buspirone Ciclosporin Clarithromycin Dexamethasone Diazepam Diltiazem Domperidone Erythromycin Estradiol Felodipine Fentanyl Finasteride Hydrocortisone Indinavir Lercanidipine Methadone Nelfinavir Nifedipine Progesterone Ritonavir Saquinavir Sildenafil |
| Page 91 of 92 |
|
Simvastatin Tacrolimus Testosterone Verapamil R-Warfarin |
||
| CYP2D6 |
Amitriptyline Carvedilol Chlorphenamine Chlorpromazine Clomipramine Codeine Dextromethorphan Donepezil Duloxetine Fluoxetine Haloperidol Imipramine Metoclopramide Metoprolol Ondansetron Oxycodone Paroxetine Propranolol Tamoxifen Timolol Tramadol Venlafaxine |
|
| CYP2C9 |
Celecoxib Diazepam Diclofenac Fluoxetine |
| Page 92 of 92 |
|
Fluvastatin Glibenclamide Glimepiride Glipizide Ibuprofen Irbesartan Losartan Meloxicam Naproxen Phenytoin S-Warfarin |
||
| CYP2C19 |
Amitriptyline Citalopram Clopidogrel Diazepam Lansoprazole Omeprazole Pantoprazole Proguanil Propanolol R-Warfarin |
|
| CYP1A2 |
Amitriptyline Clomipramine Clozapine Imipramine Theophylline R-Warfarin Caffeine |